2024年衰老生物标志物盘点01:《Cell》2023年最新发表的12大衰老生物标志物
说明:本文内容来源于权威文献、科普文章,二区健康(EequHealth.com)进行全网中英文大量内容的重新阅读、脉络梳理。同时我们对所有内容参考保留来源注明出处,方便读者考究。
版权:本文原始科研成果归属原作者,二区健康对本文具有版权,如需转载辛苦保留内容出处。
免责:二区健康对本文提到的抗衰老策略只做科普探讨,不对任何私自用作医学治疗的效果承诺和风险担责,特此免责声明。

文献参考:López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243-278. https://doi.org/10.1016/j.cell.2022.11.001
第186卷第2期,2023 年 1 月 19 日,第 243-278 页
作者:
卡洛斯·洛佩斯-奥廷123,玛丽亚· 布拉斯科 4,琳达·帕特里奇56,曼努埃尔·塞拉诺789,吉多·克罗默(Guido Kroemer)
作者单位:
1生物和生物学分子系,肿瘤大学研究所 (IUOPA),奥维耶多大学,奥维多西班牙
2阿斯图里亚斯公立医院调查研究所 (ISPA),西班牙奥维耶多
3红色癌症生物医学研究中心 (CIBERONC),西班牙马德里
4端粒和端粒酶组,分子肿瘤学项目,西班牙国家癌症中心 (CNIO),马德里,西班牙
5伦敦大学学院健康老龄化研究所遗传学、进化与环境系,英国伦敦
6马克斯·普朗克衰老生物学研究所,科隆德国
7生物医学研究所(IRB巴塞罗那),巴塞罗那科学技术研究所(BIST),巴塞罗那,西班牙
8加泰罗尼亚研究与高级研究所 (ICREA),西班牙巴塞罗那
9Altos 实验室,英国剑桥
10科德利耶研究中心、抗癌联盟设备标签、巴黎大学、索邦大学、INSERM U1138、法国大学研究所,巴黎,法国
11代谢组学和细胞生物学平台,Gustave Roussy,维勒瑞夫,法国
12巴黎癌症研究所 CARPEM,欧洲医院乔治蓬皮杜生物系,AP-HP,法国巴黎
概括
衰老是由满足以下三个前提的标志驱动的:(1)与年龄相关的表现,(2)通过实验强调它们而加速衰老,以及(3)通过治疗干预来减缓、停止或逆转衰老的机会他们。我们提出了以下十二个衰老标志:基因组不稳定、端粒磨损、表观遗传改变、蛋白质稳态丧失、巨自噬功能障碍、营养感应失调、线粒体功能障碍、细胞衰老、干细胞耗竭、细胞间通讯改变、慢性炎症和生态失调。这些特征相互关联,也与最近提出的健康特征相互关联,其中包括空间划分的组织特征、体内平衡的维持以及对压力的充分反应。
介绍
衰老研究探讨了成年期间有机体功能的衰退。自2013年第一版《衰老标志》在《细胞》杂志发表以来,已发表了近30万篇有关这一主题的文章,这一数字与上个世纪的数量相同。因此,结合十年前获得的主要知识来编写新版衰老标志的时机已经成熟。
“标志”之间的区别本质上是分散的,因为它们相互作用并且彼此不是独立的。因此,它们的分类不可避免地是任意的,但我们提出了必须适用于每个衰老标志的三个标准:(1)伴随衰老过程的变化的时间依赖性表现,(2)通过实验强调标志来加速衰老的可能性,以及(最决定性的)(3)通过针对标志的治疗干预来减缓、停止或逆转衰老的机会。我们不应该详细阐述与年龄相关的变化的概要,而应该关注从机械上解释其表现的分子、细胞和系统过程。也就是说,无论是在实验动物中还是在人类医学中,影响衰老机体的形态和功能衰退的客观量化对于测量生物衰老至关重要。事实上,生物年龄和实际年龄之间的差异可以反映年龄加速或减速操作的有效性,这些操作可以评估给定标志对衰老过程的贡献。因此,标准化的生理测量(例如,测量基础和最大能量消耗的呼吸测量法)、功能测试(例如,感觉、精神运动和认知水平)以及更加复杂的“组学”技术(例如,基因组学、表观基因组学)、转录组学、蛋白质组学和代谢组学)通常应用于单细胞水平,有助于评估健康退化的时空模式和抗衰老策略的(无效)效果。
2013年,我们提出了衰老的九个分子、细胞和系统标志:DNA不稳定、端粒磨损、表观遗传改变、蛋白质稳态丧失、营养感应失调、线粒体功能障碍、细胞衰老、干细胞耗竭和细胞间通讯改变。1最近的研究证实并扩展了所有这些特征的重要性。它们经受住了数以万计的老年研究人员的仔细审查,但它们需要更新以应对过去十年的发现。例如,2013 年,许多关于抗衰老干预措施的证据仅限于非哺乳动物模型生物,包括酵母、线虫和果蝇。幸运的是,涉及小鼠(有时还涉及非人类灵长类动物)的实验现已证实了哺乳动物中大多数这些特征的有效性。值得注意的是,当人类与年龄相关的疾病与相同的标志而不是不同的标志有因果关系时,它们同时发生和共享基因组特征的机会在统计上更高, 2临床验证了我们选择的方法。
除了对以前的标志进行必要的更新外,我们还进行了一些重组,包括以下三个额外的衰老标志:巨自噬功能丧失、慢性炎症和生态失调。巨自噬功能障碍最初被视为蛋白质稳态丧失的特殊情况。然而,巨自噬不仅影响蛋白质,还可以靶向整个细胞器和非蛋白质大分子,这证明了将其作为一个单独的实体进行讨论是合理的。此外,我们认为我们在 2013 年列出的最后一个标志,即细胞间通讯的改变,范围太大,需要单独讨论慢性炎症和与年龄相关的生态失调(图 1)。
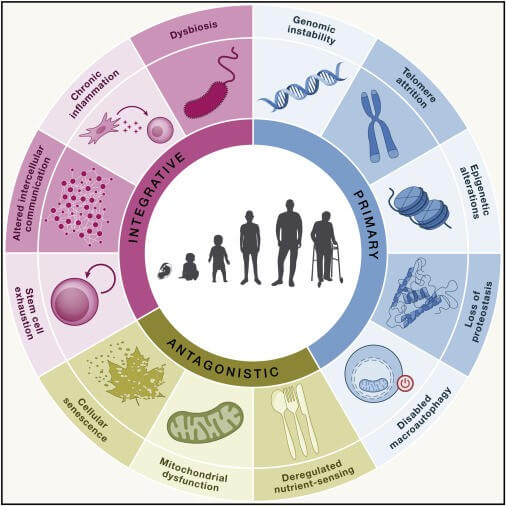
图1。衰老的特征
该方案汇集了这项工作中提出的 12 个衰老标志:基因组不稳定、端粒磨损、表观遗传改变、蛋白质稳态丧失、巨自噬功能障碍、营养感应失调、线粒体功能障碍、细胞衰老、干细胞耗竭、细胞间通讯改变、慢性炎症和生态失调。这些特征分为三类:原发性、对抗性和综合性。
老化特征的相互依赖性意味着一种特定特征的实验增强或减弱通常也会影响其他特征。这强调了这样一个事实:衰老是一个复杂的过程,必须作为一个整体来看待。因此,每个特征都应被视为未来探索衰老过程以及开发新抗衰老药物的切入点。
二区抗衰老研究院针对衰老生物标志物概要如下:
衰老是一个受多种因素影响的复杂过程。
主要原因(损坏原因):
- 基因组不稳定性:随着年龄的增长,不稳定性会增加。我们的遗传物质的突变、重排和变化的速度在增加。这种不稳定性会导致 DNA 复制/修复错误的风险升高,从而导致包括癌症在内的各种健康问题。
- 端粒缩短:随着每次细胞分裂,端粒自然缩短。最终,当它们变得太短时,细胞就会停止分裂或衰老。这会导致组织功能破坏并导致许多与年龄相关的疾病。
- 表观遗传改变:附着在 DNA 上的化学标记结构的变化可能导致基因表达异常,而不会改变遗传密码。这个过程解释了基因如何打开或关闭。
- 蛋白质稳态丧失:细胞内无法维持正确的蛋白质折叠、组装和降解。当这种平衡受到干扰时,就会导致细胞和器官水平功能障碍,例如阿尔茨海默病或帕金森病。
拮抗原因(对损害的反应):
- 营养感应失调:我们的身体检测和响应葡萄糖、氨基酸或脂肪酸等营养物质的能力受到干扰。这种情况可能导致代谢功能障碍,这是糖尿病和肥胖等疾病的主要原因,并对表观遗传学产生负面影响。
- 线粒体功能障碍:我们细胞的发电站(线粒体)功能障碍是指产生的能量太少。这会对全身的器官和系统产生负面影响,从而导致有害副产品的积累。
- 细胞衰老:这种状态是我们的细胞停止分裂并进入永久性细胞周期停滞(僵尸细胞)并导致炎症。它通常是对压力、毒素暴露、基因损伤等的反应。
老化标志的表现:
- 干细胞衰竭:干细胞的再生能力随着时间的推移而下降。这限制了组织和器官的修复。
- 细胞通讯改变:当我们的身体衰老时,细胞之间的信号通路会发生变化,这可能会导致细胞功能异常、疾病发展或体内对外部刺激的不当反应。
- 慢性炎症:慢性、低度、全身性炎症在动脉粥样硬化性心脏病、癌症和神经退行性疾病(例如阿尔茨海默病)的发展中起着关键作用。
- 巨自噬失能:巨自噬是一种细胞过程,负责去除和回收受损的细胞成分。当这个过程被破坏时,可能会导致细胞内废物积累,可能导致疾病,包括神经退行性疾病和代谢紊乱。
- 微生物群生态失调:天然微生物群落失衡、有益微生物减少、有害微生物过度生长,导致大脑功能障碍、免疫健康状况不佳和慢性炎症。
详细12个衰老生物标志物介绍如下:
1、基因组不稳定性
基因组完整性和稳定性普遍受到外源化学、物理和生物因素以及内源性挑战(例如DNA 复制错误、染色体分离缺陷、氧化过程和自发水解反应)的威胁。由这些外在或内在的损伤来源引起的广泛的遗传损伤包括点突变、缺失、易位、端粒缩短、单链和双链断裂、染色体重排、核结构缺陷以及由整合引起的基因破坏。病毒或转座子。所有这些分子改变和由此产生的基因组嵌合可能导致正常和病理性衰老。3因此,生物体进化出了一系列复杂的DNA 修复和维护机制,以应对核 DNA 和线粒体DNA (mtDNA) 造成的损伤,并确保适当的染色体结构和稳定性。这些DNA 修复网络会随着年龄的增长而失去效率,从而加剧基因组损伤的积累和细胞质4中 DNA 的异位积累(图 2 A)。
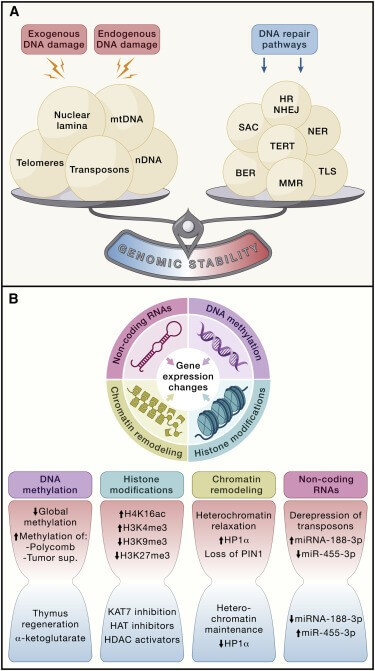
图2 .基因组不稳定、端粒磨损和表观遗传改变导致细胞完整性丧失
(A) 内源性或外源性因素会导致各种 DNA 损伤,从而导致正常和病理性衰老。这种损伤可以通过多种机制来修复,但随着年龄的增长,这些机制会失去效率。过度的 DNA 损伤、DNA 修复不足、核结构的改变和端粒磨损都有利于衰老过程。 BER,碱基切除修复; HR,同源重组; NER,核苷酸切除修复;NHEJ,非同源末端连接; MMR,错配修复; SAC,主轴装配检查点; TERT,端粒酶逆转录酶; TLS,跨损伤合成。
(B) DNA 或组蛋白乙酰化和甲基化的变化,以及染色质相关蛋白或非编码 RNA (ncRNA) 水平或活性的变化,会诱导表观遗传变化,从而导致衰老过程。沙漏的红色部分表示与年龄相关的变化,蓝色部分表示避免这些变化的策略。
核DNA
来自老年人和模型生物的细胞在核 DNA 处积累体细胞突变。 5其他形式的损伤,例如染色体非整倍性和拷贝数变异,也与衰老有关。所有这些 DNA 改变都可能影响必需基因和转录途径,导致细胞功能失调,最终可能损害组织和有机体的稳态。当 DNA 损伤影响干细胞、阻碍其在组织更新中的作用或导致其衰竭时,这一点尤其重要,从而促进衰老并增加对与年龄相关的病理的易感性。6 , 7组织学正常的人体组织中的突变负担是显着的。例如,年轻个体的正常食管上皮细胞已经表现出数百个突变,到中年时每个细胞可能携带超过 2,000 个突变。8由于完全修复由外源性和内源性挑战引起的所有基因组损伤需要消耗过多的能量,因此整个生命过程中DNA 突变的积累可能是可以容忍的。因此,细胞更注重生存而不是基因组完整性。9这些数据还表明,与致癌作用类似,仅驱动突变可能不足以加速衰老,因为它们需要由非诱变促进因子创造的宽松微环境才能渗透。 10
对哺乳动物物种突变景观的比较分析表明,物种特异性体细胞突变率与寿命呈负相关。11迄今为止,尚无明确证据表明正常的突变固定率会导致衰老,但大量研究表明DNA 修复缺陷有可能导致衰老。因此,DNA 修复机制的改变会加速小鼠的衰老,并成为多种人类早衰综合症的根源。12相反,过度表达有丝分裂检查点激酶 BubR1 的转基因小鼠表现出更长的健康寿命13(表 1)。此外,对人类和其他长寿物种的研究表明,增强的 DNA 修复机制与寿命的延长共同进化。 14 Sirtuin-6 (SIRT6) 可能在不同物种修复效率的差异中发挥着重要作用。 SIRT6 在小鼠体内的过度表达可降低基因组不稳定性,改善双链断裂修复,并延长寿命15(表 1),尽管有人提出了其他解释,例如改善葡萄糖代谢和恢复能量稳态,来解释 SIRT6 的长寿效应。16值得注意的是,最近的研究表明,8-氧代鸟嘌呤DNA 糖基化酶1 的小分子激活可增强氧化 DNA 损伤修复,并可能在衰老和与过度氧化损伤相关的其他过程中具有治疗应用。17这些发现表明,旨在减少核 DNA突变负荷或增强或改变其修复机制的干预措施可能会延缓衰老和与年龄相关的疾病的发生,但仍缺乏这方面的进一步因果证据。
表格1。针对哺乳动物的标志性干预措施的抗衰老效果示例
| 霍尔马克 | 种类/型号 | 干涉 | 结果 | 参考号 |
|---|---|---|---|---|
| 基因组不稳定性 | 老鼠 | BubR1 过度表达 | 延长寿命 | 诺斯等人。13 |
| 老鼠 | SIRT6 过表达 | 延长寿命 | 田等人。15 | |
| HGPS 鼠标 HGPS 人类 | 法尼基转移酶抑制剂 | 健康寿命和寿命延长 | 戈登等人。18 | |
| 端粒磨损 | 老鼠 | 端粒延长 | 延长寿命 | 托马斯-洛巴等人。19 |
| 端粒酶缺失小鼠 | 端粒酶重新激活 | 延长寿命 | 贾斯克利奥夫等人。20 | |
| 老鼠 | 端粒酶的药理学或遗传激活 | 延缓衰老 | 伯纳德·德·赫苏斯等人。21 | |
| 老鼠 | 超长端粒 | 延长寿命;代谢健康改善 | 穆尼奥斯·洛伦特等人。22 | |
| 老鼠 | 成年神经元的端粒维持 | 保留神经元存活和认知功能 | 希姆等人。23 | |
| 老鼠 | 基因治疗策略激活端粒酶 | 肺纤维化和再生障碍性贫血模型的改善 | 波韦达诺等人。24和 Bär 等人。25 | |
| 表观遗传改变 | 人类 | α-酮戊二酸 | 表观遗传时钟延迟 | 德米登科等人。26 |
| 人类 | GH + DHEA + 二甲双胍促进胸腺再生 | 表观遗传时钟延迟 | 法伊等人。27 | |
| 老鼠 | Kat7失活 | 延长寿命 | 王等人。28 | |
| 人类干细胞 | KAT7失活 | H3 乙酰化减少,细胞衰老减少 | 王等人。28 | |
| 老鼠 | Sirt1过度表达 | 改善基因组稳定性和代谢 | 巴特和蒂瓦里29 | |
| 老鼠 | Sirt3过度表达 | HSC再生能力逆转 | 巴特和蒂瓦里29 | |
| 老鼠 | miR-188 耗竭 | 缓解与年龄相关的血管问题 | 他等人。30 | |
| 老鼠 | miR-455-3p 过表达 | 改善线粒体和认知功能;延长使用寿命 | 库马尔等人。31 | |
| 老化或Sirt6 −/−鼠标 | 核苷逆转录酶抑制剂 | 改善健康状况和寿命 | 西蒙等人。32 | |
| 蛋白质稳态丧失 | 老鼠 | LAMP2a 在肝细胞或 HSC 中的转基因表达 | 改善老年小鼠的肝细胞活力。改善 HSC 的功能和代谢特性 | 董等人。33 |
| 老鼠 | CMA的药理诱导 | 改善疾病模型中的阿尔茨海默病病理学和动脉硬化 | 马德里加尔-马图特等人。34 | |
| 老鼠 | 重组人 HSP70 鼻内给药 | 延长寿命(m),改善认知功能和蛋白酶体活性;脑脂褐质减少 | 博布科娃等人。35 | |
| 老鼠 | 4-苯基丁酸给予老年小鼠 | 减少皮质和海马体的内质网应激并改善认知 | 哈菲兹等人。36 | |
| 人类 | 胍那苯用于 ALS 患者(FDA 批准) | 延缓进展至延髓期 | 达拉·贝拉等人。37 | |
| 巨自噬失能 | 老鼠 | Atg5的转基因过表达 | 延长寿命、代谢健康和运动功能 | Pyo 等人。38 |
| 老鼠 | beclin 1 ( Becn1 F121A/F121A )突变以减少 Bcl-2 的抑制 | 延长 C57BL/6 小鼠和早衰 klotho-ko 小鼠的寿命。延长的神经发生 | 费尔南德斯等人。39和王等人。40 | |
| 老鼠 | 饮用水中的亚精胺 | 延长寿命、减少心脏老化和氧化应激、肝窦扩张 | 艾森伯格等人。41 | |
| 老鼠 | 水杨酸盐(水杨酸盐、乙酰水杨酸盐) | EP300抑制;自噬依赖性保肝作用;改善癌症免疫监视 | 卡斯托尔迪等人。42 | |
| 老鼠 | 去甲二氢愈创木酸 | EP300 抑制可延长寿命 (m) | 特齐尔等人。43 | |
| 人类 | 糖尿病前期女性口服 NMN(III 期试验) | 骨骼肌的胰岛素敏感性增加 | 吉野等人。44 | |
| 人类 | 帕金森病患者的口服 NR(1 期试验) | 临床改善,血清和脑脊液中炎症细胞因子减少 | 布雷克达尔等人。45 | |
| 人类 | 过去 5 年内患有 2 种非黑色素瘤皮肤癌的患者进行 NAM(3 期试验) | 降低新发非黑色素瘤皮肤癌和角化病的发生率 | 陈等人。46 | |
| 人类 | 尿石素 A 适用于中年成人(随机第 2 期) | 提高有氧耐力和身体表现;血浆CRP降低 | 辛格等人。47 | |
| 营养感应失调 | 老鼠 | 6 个月时诱导型 GH 受体敲除 | 长寿和增强胰岛素敏感性;肿瘤较少(m) | 杜兰-奥尔蒂斯等人。48 |
| 老鼠 | 30% 的雄性 C57BL/6J 小鼠热量限制 | 寿命延长 10%–30% | 阿科斯塔-罗德里格斯等人。49 | |
| 人类 | 2 年热量限制 14%(第 2 阶段) | 改善胸腺生成和对脂肪组织的抗炎作用。 PLA2G7 表达减少(小鼠基因敲除可改善代谢健康) | 斯帕达罗等人。50 | |
| 老鼠 | 饮用水中的β-羟基丁酸盐 | 更高的能量消耗,改善运动健康和记忆力 | 范等人。51 | |
| 线粒体功能障碍 | 老鼠 | TPP-噻唑(呼吸链复合物 IV 抑制剂) | 改善老年小鼠的线粒体代谢、减少内脏脂肪和提高葡萄糖耐量 | 塔瓦莱等人。52 |
| 老鼠 | CRMP 优先作用于解偶联肝细胞线粒体 | 对肥胖老年小鼠给药可减少肝脂肪变性和肝脏胰岛素抵抗 | 戈德克等人。53 | |
| 老鼠 | 埃拉米普肽抑制线粒体通透性转变 | 改善线粒体功能并避免舒张性心功能不全,特别是与 NMN 联合使用 | 张等人。54 | |
| 灵长类动物 | 用 CRMP 治疗食蟹猴和恒河猴自发性或饮食引起的肥胖 | 增强肝脏线粒体脂肪氧化,改善胰岛素耐受性,降低肝脏和血浆甘油三酯,降低胆固醇 | 戈德克等人。55 | |
| 人类 | 评估艾拉普肽治疗巴斯综合征的临床试验(随机 2/3 期试验) | 改善步行测试、肌肉和心脏参数,整体改善 | 里德·汤普森等人。56 | |
| 人类 | 老年人补充左旋肉碱的临床试验(二期试验) | 运动期间增加肌肉肉碱含量和脂肪酸氧化 | 奇等人。57 | |
| 细胞衰老 | 老鼠 | p16 表达细胞的基因消融 | 增加健康和寿命。治疗从1岁开始 | 贝克等人。58 |
| 老鼠 | 达沙替尼 + 槲皮素的 senolytic 治疗 | 增加健康和寿命。治疗从2岁开始 | 徐等人。59 | |
| 老鼠 | 使用非瑟酮进行 senolytic 治疗 | 增加健康和寿命。治疗从1.6岁开始 | 尤瑟夫扎德等人。60 | |
| 人类 | 达沙替尼 + 槲皮素对肺纤维化患者进行 senolytic 治疗(1 期试验) | 改善身体表现;血清中促炎和促纤维化因子减少;尿液中αKlotho升高 | 正义等人。61 | |
| 人类 | 达沙替尼 + 槲皮素对糖尿病肾病患者进行 senolytic 治疗(1 期试验) | 减少脂肪组织中的衰老细胞和巨噬细胞以及血清中的促炎因子 | 希克森等人。62 | |
| 干细胞衰竭 | 老鼠 | OSKM的转基因表达 | HGPS 早衰小鼠的寿命延长。正常小鼠海马神经发生和功能的保存。受伤后或随后受伤后立即改善组织修复 | Wang 等人,28 Ocampo 等人,63 Browder 等人,64 Chen 等人,65 Hishida 等人,66 Rodríguez-Matellán 等人,67 Gau 等人,68和Doeser 等人。69 |
| 老鼠 | AAV2驱动的OSK在眼中的表达 | 恢复老年小鼠和患有青光眼的小鼠的视力。改善受损视神经的修复 | 卢等人。70 | |
| 改变细胞间通讯 | 老鼠 | 用盐水/白蛋白稀释年老小鼠的血液 | 多个组织的年轻化 | 梅赫迪普尔等人。71 |
| 老鼠 | 输血 | 改善肌肉修复;减少肝脏脂肪变性和纤维化 | 雷博等人。72 | |
| 老鼠 | 人脐带血浆 | 改善海马神经发生 | 卡斯特拉诺等人。73 | |
| 老鼠 | 异时性联体共生 | 多个组织的年轻化 | 马等人。74和帕洛维奇等人。75 | |
| 老鼠 | CCL3/MIP1α 给药 | HSC 年轻化 | 马等人。74 | |
| 老鼠 | TIMP2 静脉注射 | 海马回春 | 卡斯特拉诺等人。73 | |
| 老鼠 | 将IL-37注射到年老小鼠体内 | 改善新陈代谢和耐力运动 | 巴拉克等人。76 | |
| 老鼠 | GDF11 静脉注射给药 | 使大脑、肌肉和胰腺恢复活力,但具有促纤维化作用 | 弗罗利希和文奇格拉77 | |
| 老鼠 | VEGF转基因过度表达 | 改善健康和寿命,增强肝脏和肌肉修复 | 格鲁内瓦尔德等人。78 | |
| 小鼠人类细胞 | 雅普表达 | 使衰老细胞恢复活力,预防衰老特征的出现 | 斯拉迪切克-马滕斯等人。79 | |
| 人体细胞 | 来自年轻成纤维细胞的 ECM | 老化衰老细胞的复兴 | 崔等人。80 | |
| 老鼠 | 软骨素 6-磺基转移酶过度表达 | 改善老年小鼠的记忆力 | 杨等人。81 | |
| 慢性炎症 | 老鼠 | 在 16 至 18 月龄的 C57BL/6 小鼠中用依那西普阻断 TNF-α | 预防肌肉减少症和延长寿命 (f) | 德斯丁-米科等人。82;夏奥拉蒂等人。83 |
| 鼠 | 24至26月龄雄性Wistar白化大鼠中使用依那西普阻断TNF-α | 预防认知缺陷、内皮功能障碍、外周和神经炎症 | 戈克梅兹等人。84 | |
| 老鼠 | 敲除骨髓细胞中的前列腺素 E 2受体 EP2 或用 EP300 抑制剂治疗 C57BL/6 小鼠 | 改善认知并减少与年龄相关的炎症 | 米哈斯等人。85 | |
| 老鼠 | C57BL/6 小鼠中 NLRP3 的敲除 | 由于卵巢老化减少,葡萄糖耐量、认知能力、运动表现和女性生育能力得到改善 | 马林-阿吉拉尔等人。86 | |
| 人类 | 用卡那单抗治疗有心肌梗塞病史且高 hsCRP 的患者(3 期试验) | 降低高血压和糖尿病的发病率;降低复发性心肌梗塞和非小细胞肺癌的频率 | 里德克等人。87 | |
| 生态失调 | HGPS鼠标 | WT小鼠粪便微生物群移植;嗜粘蛋白阿克曼氏菌给药 | 延长健康寿命和寿命 | 巴尔塞纳等人。88 |
| SAMP8鼠标 | 植物乳杆菌GKM3 | 促进长寿和减轻与年龄相关的认知障碍 | 林等人。89 | |
| 老鼠 | 将年轻小鼠的微生物群移植到老年宿主身上 | 改善大脑健康和免疫力的维持 | 博梅等人。90 | |
| 老鼠 | 粪便微生物移植 | 改善老年小鼠的卵巢功能 | 徐等人。91 | |
| 老鼠 | 粪便微生物移植 | 改善淋巴结生发中心反应 | 斯特贝格等人。92 | |
| 老鼠 | 吲哚代谢物 | 减少衰老过程中的炎症 | 克里希南等人。93 | |
| 老鼠 | 短链脂肪酸 | 恢复老年小鼠的小胶质细胞功能 | 克莱恩等人。94 | |
| 人类 | 口服Akkermansia muciniphila(随机 1/2 期试验) | 改善肥胖或糖尿病患者的代谢参数 | 德波米尔等人。95 |
线粒体DNA
影响 mtDNA 的基因组不稳定性可能会导致衰老和与年龄相关的病理。96 mtDNA 由于其复制指数高、修复机制效率有限、氧化微环境以及缺乏包含这种小 DNA 分子的保护性组蛋白,因此受到与衰老相关的突变和缺失的强烈影响。在衰老过程中,人体组织的体细胞 mtDNA 改变会增加,但目前尚不清楚这种增加是否真正在功能水平上影响衰老过程。由于“异质性”,mtDNA 突变导致衰老的因果关系很难评估,“异质性”意味着同一细胞内突变基因组和野生型基因组共存。然而,对老化细胞的深度测序表明,它们的 mtDNA突变负荷可能通过克隆扩增事件大幅增加。97在灵长类卵母细胞和体细胞组织中也观察到线粒体突变随着年龄的增长而加速扩展, 98以及神经退行性疾病患者的淋巴母细胞中也观察到了这一现象。99值得注意的是,超灵敏测序表明,衰老细胞中的大多数 mtDNA 突变是由mtDNA 聚合酶γ引起的复制错误引起的,而不是氧化应激引起的。96
由 mtDNA 损伤和部分表型衰老引起的人类疾病提供了 mtDNA 突变可能直接参与衰老和年龄相关病理的初步证据。100对缺乏 DNA 聚合酶 γ 的小鼠进行的研究得出了进一步的致病证据,这些小鼠表现出与mtDNA缺失而不是点突变相关的加速衰老和寿命缩短101(表 1)。总体而言,这些数据表明,线粒体 DNA 突变的避免、减弱或纠正可能有助于延长健康寿命和寿命。然而,与核DNA 突变的情况一样,通过线粒体 DNA 修复机制功能的增强来减缓衰老的实验证据仍然很大程度上缺失。
核架构
核纤层构成了束缚染色质和蛋白质复合物的支架,其缺陷可能会导致基因组不稳定。102加速衰老综合征,如 Hutchinson-Gilford 和 Néstor-Guillermo早衰综合征(分别为 HGPS 和 NGPS)是由编码核纤层蛋白成分的基因LMNA和BANF1突变引起的。核纤层的改变和称为早老素的异常前核纤层蛋白 A亚型的产生也是正常人类衰老的特征,并且核纤层蛋白B1 水平在细胞衰老过程中下降。18动物和细胞模型有助于识别衰老和早衰引起的核层畸变引起的反应机制和应激途径,包括肿瘤抑制蛋白p53 (TP53) 的激活、生长激素轴的失调以及成体干细胞的损耗。18
在 HGPS 小鼠模型中,降低 prelamin A 或早老蛋白水平可延迟早衰特征的出现并延长寿命,这一观察结果证实了核纤层异常与过早衰老的因果关系。这可以通过全身注射反义寡核苷酸、法呢基转移酶抑制剂、他汀类药物和氨基二磷酸盐的组合、恢复生长激素轴或阻断 NF-κB 信号传导来实现。 103其中一些干预措施已被批准用于早衰患者。 104此外,最近开发了基因编辑策略来纠正HGPS 患者细胞和该疾病动物模型中的LMNA突变。 105 , 106希望这些方法能够在临床上应用于未来的早衰症治疗,但迄今为止,没有证据表明减少早衰素会延缓正常衰老。
2、端粒磨损
染色体末端(端粒)的 DNA 损伤会导致衰老和与年龄相关的疾病。107复制DNA 聚合酶无法完成真核 DNA 端粒区域的复制。因此,经过几轮细胞分裂后,端粒经历大幅缩短,导致基因组不稳定,最终导致细胞凋亡或细胞衰老。这些有害影响可以通过端粒酶的逆转录酶活性来预防,端粒酶是一种活性核糖核蛋白,可以延长端粒以维持其足够的长度。108 , 109然而,大多数哺乳动物体细胞不表达端粒酶,这导致整个生命过程中染色体末端端粒序列逐渐和累积的侵蚀。有几个例子表明,端粒磨损通过限制恶性细胞的复制寿命来减弱癌变。因此,与明确有利于肿瘤发生的基因组不稳定性相反,端粒磨损可能会对抗恶性肿瘤。因此,我们认为端粒磨损是衰老的一个标志,与基因组不稳定是分开的。110
人类端粒酶缺乏与肺纤维化、再生障碍性贫血和先天性角化不良等疾病的过早发展有关,所有这些都会阻碍受影响组织的再生能力。111在许多不同物种(包括人类和小鼠)的正常衰老过程中也观察到端粒缩短。 112端粒损耗率受年龄、遗传变异、生活方式和社会因素的影响;取决于受影响细胞的增殖活性;并预测多种物种的寿命。112端粒脱帽也可能是由于庇护蛋白的缺陷造成的,庇护蛋白是一组阻止染色体末端DNA 损伤反应并调节端粒长度的蛋白质。几种庇护蛋白成分的功能丧失模型表明,即使存在正常长度的端粒,组织再生能力也会下降并加速衰老。113
转基因动物模型揭示了端粒磨损、细胞衰老和机体衰老之间的因果关系。端粒缩短或延长的小鼠分别表现出寿命缩短或延长。19值得注意的是,当端粒酶通过基因重新激活时,端粒酶缺陷小鼠的过早衰老可以得到恢复20(表 1)。此外,端粒酶的药物激活或全身病毒转导可以延缓小鼠的正常衰老21 ,而端粒超长的小鼠则显示出寿命延长和代谢健康改善22(表1)。同样,在阿尔茨海默氏病模型中,经过改造以维持成年神经元端粒酶生理水平的小鼠可以保留这些细胞的存活并维持认知功能23(表1)。因此,衰老可以通过端粒酶激活来调节。
端粒酶激活可延缓衰老并治疗端粒疾病
在人类中,许多研究提供了端粒长度短与年龄相关疾病之间因果关系的证据。 114特别是,具有短端粒的小鼠模型的产生已证明,端粒磨损是端粒综合征115和流行的年龄相关疾病(例如肺纤维化和肾纤维化)的根源。 24 , 116端粒动力学和机体衰老之间的这些联系催生了新的干预措施的设计,以延缓衰老和与年龄相关的疾病。例如,使用基因治疗策略激活端粒酶已显示出对肺纤维化和再生障碍性贫血小鼠模型的治疗效果。24 , 25
3、表观遗传改变
导致衰老的多种表观遗传变化包括DNA 甲基化模式的改变、组蛋白翻译后修饰异常、染色质重塑异常以及非编码RNA (ncRNA) 功能失调(图 2 B)。这些调节性且通常是可逆的变化会影响基因表达和其他细胞过程,导致多种与年龄相关的人类病理的发生和进展,例如癌症、神经退行性变、代谢综合征和骨病。大量的酶系统参与表观遗传模式的产生和维持。这些酶包括DNA 甲基转移酶、组蛋白乙酰化酶、脱乙酰酶、甲基化酶和去甲基化酶,以及参与染色质重塑或ncRNA合成和成熟的蛋白质复合物。
DNA甲基化
随着时间的推移,人类DNA 甲基化景观积累了多种变化。 117早期研究描述了与年龄相关的整体低甲基化,但进一步的分析表明,特定基因座,包括几个肿瘤抑制基因和 Polycomb 靶基因的基因座,随着年龄的增长而高度甲基化。来自患有早衰综合症的患者和小鼠的细胞也表现出DNA 甲基化变化,部分重现了正常衰老过程中发现的变化。118大多数与年龄相关的表突变的功能后果是不确定的,因为大多数变化影响内含子和基因间区域。119
基于选定位点 DNA 甲基化状态的表观遗传时钟已被引入,以预测实际年龄和死亡风险,并评估可能延长人类寿命的干预措施。 119这一点已通过旨在胸腺再生的方案得到证实,该方案改善了许多与年龄相关的疾病的风险指数,并且治疗 1 年后平均表观遗传年龄比基线低约 1.5 岁。此外,对人类发病率和死亡率的预测显示,表观遗传年龄与实际年龄相比减少了 2 年,这种情况在停止治疗后持续了 6 个月。27同样,补充 7 个月的 α-酮戊二酸使表观遗传时钟倒退 8 年。 26总之,DNA 甲基化变化与衰老相关,但没有明确的证据表明它们确实导致衰老。需要进一步的研究来证明 DNA 甲基化维持缺陷会导致加速衰老,而 DNA 甲基化模式维持保真度的提高可以延长寿命。还需要确定负责调节老年人类甲基化组中发生的变化的分子驱动因素。
组蛋白修饰
组蛋白的整体丢失及其翻译后修饰的组织依赖性变化也与衰老密切相关。组蛋白表达增加可延长果蝇的寿命,120而在老年人和早衰患者的成纤维细胞中发现组蛋白 H4K16乙酰化或 H3K4 三甲基化水平增加以及 H3K9 或 H3K27 三甲基化水平降低。这些组蛋白修饰可导致转录变化、细胞稳态丧失以及与年龄相关的代谢下降。121值得注意的是,端粒异色标记的丢失已被证明会导致端粒延长。122
组蛋白去甲基酶通过靶向关键长寿途径的成分(例如胰岛素/胰岛素生长因子-1 (IGF-1)信号通路)来调节寿命。其他组蛋白修饰酶,例如 SIRT 蛋白脱乙酰酶家族成员和 ADP-核糖基转移酶,也有助于健康衰老。29 SIRT1的转基因过度表达可改善小鼠衰老过程中的基因组稳定性和代谢效率,但不会延长寿命。29线粒体SIRT3的过度表达可逆转衰老造血干细胞(HSC)丧失的再生能力,并可介导饮食限制对长寿的有益影响。123同样,小鼠中的Sirt6消除会导致加速衰老,124而Sirt6过度表达可延长寿命。16其根本机制源于这样一个事实:Sirt6 是一种多任务蛋白,能够将染色质动力学与代谢和DNA 修复相互关联。125最后,Sirt7缺乏会导致整体基因组不稳定、代谢功能障碍和过早衰老。29总之,这些发现与脱乙酰酶活性降低会导致染色质松弛、DNA 损伤剂暴露增加以及基因组不稳定性增强的观点相一致。126相反,人类干细胞中组蛋白乙酰转移酶KAT7的基因失活会降低组蛋白 H3K14乙酰化并减轻细胞衰老特征。28此外,静脉注射编码 Cas9/sg-Kat7 的慢病毒载体可改善正常小鼠和早衰小鼠的肝细胞衰老和肝脏老化,并延长寿命。28组蛋白乙酰转移酶抑制剂还可改善早衰表型并延长早衰小鼠的寿命,而组蛋白脱乙酰酶激活剂则部分通过上调 SIRT1 活性来延长寿命。127总之,这些发现表明,应进一步探索组蛋白修饰剂作为对抗年龄相关认知衰退的治疗策略的一部分,尽管目前尚不清楚这些干预措施是否通过纯粹的表观遗传机制,通过影响DNA 修复和基因组来影响衰老和寿命。稳定性或通过影响代谢或信号传导途径的转录改变。
染色质重塑
除了 DNA 和组蛋白修饰剂之外,一些染色体蛋白和染色质重塑因子,例如与基因组稳定性DNA 修复有关的异染色质蛋白 1α (HP1α) 和Polycomb 族蛋白,也可能调节衰老。128这些表观遗传因素的改变会导致染色质结构的深刻变化,包括整体异染色质丢失和重新分布,这是衰老细胞中的常见事件。
这些染色质改变与衰老的因果关系已在无脊椎动物中进行了大量研究,其中 HP1α 的功能丧失突变会缩短寿命,而其过度表达则会延长健康寿命和寿命129(表 1 )。在哺乳动物中进行的类似研究仍然有限,但大多数研究表明异染色质松弛会导致衰老和衰老相关病理,而异染色质的维持可促进长寿。例如,PIN1(一种保护异染色质所必需的脯氨酰 异构酶)的丢失与从果蝇到哺乳动物等不同物种的过早衰老和神经变性有关130(表1)。然而,旨在通过增强染色质重塑因子的功能来延长脊椎动物寿命的实验仍然缺失。
非编码RNA
大量且不断增长的 ncRNA,包括 lncRNA(例如端粒 RNA 或 TERRA)、microRNA (miRNA) 和环状 RNA,已成为能够影响衰老的表观遗传因素。 ncRNA 通过转录后靶向长寿网络的组成部分或通过调节干细胞行为来调节健康寿命和寿命。131环状 RNA 介导胰岛素/IGF-1信号通路对果蝇寿命的影响, 132但大多数研究都集中在 miRNA 上,并且对于其他 ncRNA 可能在多大程度上源自转录噪音以及它们的调节仍存在争议。在人类生理学和病理学中的作用仅局限于少数特定情况。133
功能获得和丧失的研究首先证实了几种 miRNA 调节无脊椎动物寿命的能力。随后的小鼠研究为 miRNA 在衰老过程中的功能相关性提供了因果证据(表 1)。例如,衰老过程中骨骼内皮中的 miRNA-188-3p 表达上调,并导致与时间流逝相关的血管问题。小鼠体内 miR-188 的耗竭减轻了有益骨毛细血管亚型与年龄相关的下降,而该 miRNA 的内皮特异性过度表达会减少骨量并延迟骨再生。30相反,小鼠中 miR-455-3p 的缺失会对线粒体动力学、认知行为和寿命产生有害影响,而其过度表达则保留这些功能并延长寿命。31总体而言,这些发现表明 miRNA 可能会导致衰老和衰老相关病理,并代表延迟或改善这些病症的潜在治疗靶点。
反转录转座子的去抑制
最近的研究揭示了逆转录转座子在包括人类在内的复杂后生动物衰老中的作用。134这些逆转录转座元件是可移动的遗传单位,可以利用涉及 RNA 中间体的分子机制从一个基因组位置移动到另一个基因组位置。逆转录转座子由长散布核元件(LINE) 和 SINE组成,前者编码逆转录转座所需的蛋白质,后者是劫持 LINE 蛋白质机制的短非编码 RNA。逆转录转座子在衰老细胞中和一生中被重新激活,并通过遗传和表观遗传变化或通过将逆转录转座子核酸鉴定为外源DNA后触发的免疫途径激活而产生有害影响。 134从机制上讲,LINE-1 RNA 的表观遗传去抑制会抑制表观遗传读取器 Suv39H1,2,导致 H3K9me3 和异染色质整体减少, 135而LINE-1 RNA 的逆转录会产生双链 cDNA,从而激活 cGAS/STING/干扰素途径。136
使用核苷逆转录酶抑制剂(NRTI)治疗可抑制或减弱逆转录转座,延长Sirt6缺失小鼠的寿命并改善健康寿命,改善骨骼和肌肉表型(表1)。同样,用 NRTI 治疗老年野生型小鼠可降低 DNA 损伤标记物的水平。32此外,体内反义寡核苷酸靶向逆转录转座子可延长早衰小鼠的寿命。135值得注意的是,百岁老人中罕见的SIRT6变异是 LINE1逆转录转座子的更强抑制因子,增强了基因组稳定性,并且比野生型SIRT6能够更强有力地杀死癌细胞。137总的来说,这些发现表明逆转录转座子会导致衰老过程,而反对逆转录转座子活性的干预措施可能会延长健康寿命。使用针对反转录转座子不同功能的药物对老年人群进行进一步的临床研究可能会描绘出针对衰老和衰老相关病理的新干预策略。
基因表达变化
所有上述表观遗传因素影响的机制都集中在基因表达水平的调节上。衰老会导致转录噪音增加以及许多 mRNA 的异常产生和成熟。138 , 139对人类和其他物种的年轻和年老组织进行基于微阵列的比较,发现了与年龄相关的转录特征,这些转录特征是由衰老过程中发生的表观遗传变化引起的。环境暴露还会通过 DNA 甲基化改变和组蛋白修饰引起基因调控的改变,并促进与衰老相关的表观遗传变化,包括表观遗传时钟的加速。140
对小鼠整个生命周期中多个年龄段的多种细胞类型和器官的单细胞转录组学和血浆蛋白质组学揭示了衰老过程中显着的基因表达变化。 138这些变化特别影响某些生物过程,例如炎症、蛋白质折叠、细胞外基质(ECM) 调节和线粒体功能,这些过程在衰老过程中广泛失调。141衰老过程中在不同组织中观察到的常见表达模式可能有助于指导未来旨在改善健康和寿命的干预措施(表 1)。同样,在衰老过程中观察到的转录和转录后效率和保真度的下降及其对蛋白质组健康的负面影响也可能为长寿策略带来新的机会。139
4、蛋白质稳态丧失
衰老和一些与年龄相关的疾病,如肌萎缩侧索硬化症(ALS)、阿尔茨海默病、帕金森病和白内障,与蛋白质稳态或蛋白质稳态受损有关,导致错误折叠、氧化、糖化或泛素化蛋白质的积累,通常形成聚集体,如细胞内包涵体或细胞外淀粉样斑块。142
蛋白质稳态崩溃
由于错误翻译、错误折叠或不完整蛋白质的产生增加,细胞内蛋白质稳态可能被破坏(图 3)。对核糖体蛋白RPS23进行基因操作以提高 RNA 到蛋白质翻译的准确性,可延长粟酒裂殖酵母、秀丽隐杆线虫和黑腹果蝇的寿命,143而 RPS9 的突变有利于易错翻译,导致小鼠过早衰老。144导致蛋白质稳态网络崩溃的另一个机制在于蛋白质翻译伸长减慢和累积氧化损伤,从而越来越分散分子伴侣折叠细胞健康所需的健康蛋白质的注意力。145此外,许多与年龄相关的神经退行性疾病,包括 ALS 和阿尔茨海默病,都可能是由蛋白质突变引起的,这些突变使它们本质上容易错误折叠和聚集,从而使维持神经功能所需的蛋白质修复、去除和周转机制饱和。健康状态。146

图3 .蛋白质和细胞器更新的损失
蛋白质稳态丧失和巨自噬功能丧失的特点是偏离年轻平衡状态,其中废物的积累是由各种与年龄相关的变化引起的,同时废物的清除通过各种机制受到损害。列出了这些改变的功能后果。左侧和右侧举例说明了一些重建蛋白质稳态和自噬的策略。
当确保质量控制的机制失败时,蛋白质稳态网络也会崩溃,例如,由于内质网(ER)中未折叠蛋白质反应(UPR)的功能降低, 147当正确折叠蛋白质的稳定性受到损害或降解机制时蛋白酶体或溶酶体对蛋白质的吸收变得不足(图3)。在衰老的器官中观察到蛋白酶体活性降低,包括短命鱼Nothobranchius Furzeri的大脑。 148此外,一些单泛素化蛋白在果蝇、小鼠、猴子和人类的衰老组织中积累,正如组蛋白 2A所记录的那样。149
溶酶体对蛋白质的降解可以通过分子伴侣介导的自噬 (CMA) 以特定方式实现,其中暴露类似于 KFERQ 的五肽基序的蛋白质首先与热休克蛋白HSC70结合,然后与溶酶体相关膜蛋白 2A 型结合。 LAMP2A),它促进客户蛋白易位到溶酶体的腔中。150小鼠肝脏 LAMP2A 表达随着年龄的增长而下降,其转基因重新表达可减少肝脏衰老。151蛋白质聚集体在包含在双膜囊泡(自噬体)中后,也可以通过巨自噬去除,以便随后与溶酶体融合。152由于自噬体可以包裹非蛋白质结构,因此该过程将在下一个标志部分(禁用的巨自噬)中与蛋白质稳态分开讨论。尽管如此,刺激自噬构成了消除细胞内蛋白质聚集体的有效策略。153
蛋白质稳态、衰老和长寿
一般蛋白质稳态的扰动会加速衰老。例如,给黑腹果蝇喂食晚期糖基化终末产物(AGE) 或脂褐素(共价交联的蛋白质、糖和脂质的聚集体)会导致 AGE 修饰和羰基化蛋白质的积累,从而缩短健康寿命和寿命,从而导致黑腹果蝇的健康寿命和寿命缩短。敲低溶酶体蛋白酶组织蛋白酶 D后,这种作用会进一步增强。 154蛋白酶ZMPSTE24的缺失会破坏 prelamin A 的正常蛋白水解成熟,并导致小鼠出现早衰综合症,这与在具有 ZMPSTE24 功能丧失突变的人类中观察到的表型相似。18在小鼠中,敲除神经元中的 LAMP2A(CMA 必需的)会深刻影响蛋白质组,产生与阿尔茨海默病患者中发现的类似变化。事实上,抑制小鼠体内的 CMA 会加剧实验性阿尔茨海默氏病,而药理学 CMA激活剂的刺激则可减轻该病。155
实验性改善蛋白质稳态可以延缓衰老过程(表 1)。重组人 HSP70 蛋白鼻内应用到小鼠体内可增强蛋白酶体活性,降低脑脂褐素水平,增强认知功能并延长寿命。35同样,给老年小鼠施用化学伴侣4-苯基丁酸酯可以减少大脑中的内质网应激并改善认知能力。 36在线虫和果蝇中,转染强制分离的蛋白酶体亚基过度表达可改善蛋白质稳态并延长寿命。156在小鼠中,通过LAMP2a HSC转基因表达刺激 CMA可改善目标细胞群的存活率,33与 CMA 的药理增强可减轻阿尔茨海默氏病和动脉硬化的观察结果一致。155 , 34因此,CMA 的激活可能构成延迟衰老过程的有效策略。
一项 3 期临床试验表明,在最近诊断为 ALS 的患者中,给予抗高血压药物 guanabenz 可抑制进展至危及生命的延髓阶段。37胍可能会刺激真核翻译起始因子 2α (eIF2α) 的磷酸化(或抑制去磷酸化),这种现象发生在作为 UPR 一部分的“综合应激反应 (ISR)”背景下,157尽管它仍然存在关于胍那苯的作用在多大程度上是由 ISR 的刺激介导的争论。158重要的是,eIF2α 磷酸化会导致 RNA 翻译从 5′ 帽依赖性转变为 5′ 帽非依赖性RNA 翻译,因为后者可通过多种延长寿命的操作得到增强。159此外,eIF2α 磷酸化对于诱导应激颗粒至关重要,而应激颗粒是通过限制蠕虫饮食来延长寿命所必需的。160最后,eIF2α 磷酸化对于诱导自噬是不可或缺的,161这是一种主要的抗衰老机制(见下文),这表明 UPR 和自噬之间在长寿途径中存在相互影响。未来的研究必须确定胍那苯减轻神经变性的能力是否是通过 ISR 刺激或替代机制介导的。事实上,有人提出 ISR 抑制剂也可用于治疗神经退行性疾病。162
5、巨自噬失能
巨自噬(我们将称为“自噬”)涉及将细胞质物质隔离在双膜囊泡(自噬体)中,随后与溶酶体融合以消化管腔内容物。152因此,自噬不仅参与蛋白质稳态,还影响非蛋白质大分子(例如异位胞质 DNA、脂质囊泡和糖原)和整个细胞器(包括“线粒体自噬”靶向的功能失调的线粒体,以及导致“溶食”的其他细胞器) 、“网状自噬”或“pexophagy”),以及入侵病原体(“异体自噬”)。152与年龄相关的自噬下降是细胞器更新减少的最重要机制之一,证明其作为衰老新标志的讨论是合理的。需要注意的是,参与自噬过程的基因和蛋白质也参与替代降解过程,例如 LC3 相关的细胞外物质吞噬作用, 163以及以外逸球形式排出细胞内废物(例如,功能失调的线粒体)。随后被巨噬细胞清除。 164尽管如此,有强有力的证据表明自噬的核心过程与衰老有关(图 3)。
自噬抑制导致加速衰老
在人类中,自噬相关基因(例如ATG5、ATG7和BECN1 )的表达随着年龄的增长而下降。从长寿父母的后代中分离出的165 个CD4 + T 淋巴细胞与年龄匹配的对照相比显示出增强的自噬活性。 166衰老供体的循环 B 和 T 淋巴细胞中自噬的减少伴随着促自噬代谢物亚精胺的减少。167 , 168同样,在啮齿动物中,一些器官的自噬逐渐恶化,这支持了自噬通量随着年龄的增长而受到损害的观点。自噬通量的减少可能参与蛋白质聚集体和功能失调的细胞器的积累、病原体消除的减少以及炎症的增强,因为自噬消除了与炎症小体及其上游触发因素有关的蛋白质。169
自噬的基因抑制加速了模型生物体的衰老过程。这个过程是部分可逆的,如在小鼠中所示,Atg5被多西环素诱导的shRNA下调。Atg5敲低会导致多个器官系统过早退化和衰老,从而导致过早死亡。170停用多西环素后,自噬恢复伴随着全身炎症的减轻和衰老的节段性减少。值得注意的是,在该模型中,自噬的短暂抑制伴随着恶性肿瘤发病率的大幅增加。因此,自噬显然是一种肿瘤抑制机制,可能涉及细胞自主过程和癌症免疫监视。153在患者中,调节或执行自噬的基因功能丧失突变与多种心血管、感染、神经退行性、代谢、肌肉骨骼、眼部和肺部疾病有因果关系,其中许多疾病类似于早衰。组织病理学和功能水平。152 , 153
自噬刺激可延缓衰老
有充分的证据表明,刺激自噬流可以延长模型生物体的健康寿命和寿命(表 1)。例如,仅增加肠道肠细胞的自噬可以延长果蝇的寿命。120在小鼠中,在普遍表达的启动子的控制下Atg5的转基因过表达足以延长寿命并改善代谢健康和运动功能。38此外, beclin 1 ( Becn1 F121A/F121A )的敲入突变可减少 Bcl-2 对其的抑制,从而导致自噬流增加,并延长寿命。这种效应与年龄相关病理和自发肿瘤发生的减少有关,39以及神经发生的增加。40
给小鼠口服补充亚精胺可诱导多个器官的自噬,将寿命延长高达 25%,同时心脏衰老也会减少。心肌细胞特异性敲除Atg7后,后一种效应就会消失,表明它依赖于自噬。41从机制上讲,亚精胺的促自噬作用与乙酰转移酶EP300的抑制有关(导致 几种核心自噬蛋白的乙酰化减少) 171或与 eIF5A 的抑制作用有关,而 eIF5A 对自噬转录的合成至关重要因子TFEB。167在这些因素中,EP300 是延年益寿药物去甲二氢愈创木酸43和水杨酸盐的作用靶点。42 C646 对 EP300 的药理学抑制作用类似于亚精胺对自噬和癌症免疫监视的刺激作用。172当来自老年人类供体的循环 B 淋巴细胞或 CD8 + T 细胞在亚精胺存在下进行培养时,细胞的 TFEB 和 eIF5A 恢复到幼年水平,同时自噬流也恢复正常。167 , 168此外,在果蝇中,由于杂合突变或脱氧马尿苷合酶敲低导致的马尿苷缺陷,消除了通过补充亚精胺延长寿命的效果。 173小鼠 T 细胞中脱氧马尿苷合酶缺乏会引发严重的肠道炎症,并伴有表观遗传重塑和三羧酸循环的重新连接,174而野生型小鼠的亚精胺治疗可预防结肠炎和结肠癌。175因此,EP300 抑制和 eIF5A 抑制似乎都是解释亚精胺体内作用的合理目标。
诱导线粒体自噬并对小鼠健康寿命产生积极影响的药物包括 NAD +前体(如烟酰胺、烟酰胺单核苷酸和烟酰胺核苷)176和尿石素 A。177临床试验已证明 NAD +前体在化学预防非-黑色素瘤皮肤癌,46逆转糖尿病前期女性的胰岛素抵抗,44以及减少帕金森病患者的神经炎症。45此外,一项 3 期试验揭示了尿石素 A 提高肌肉力量和降低 C 反应蛋白 (CRP) 的能力。47
6、营养感应失调
营养感应网络在进化中高度保守。它包括细胞外配体,例如胰岛素和 IGF、与其相互作用的受体酪氨酸激酶,以及细胞内信号级联。这些级联涉及 PI3K-AKT 和 Ras-MEK-ERK 途径,以及转录因子,包括 FOXO 和 E26 因子,它们反式激活参与多种细胞过程的基因。雷帕霉素 (MTOR) 复合物 1 (MTORC1) 的机制靶标对营养物质(包括葡萄糖和氨基酸)以及缺氧和低能量等应激源作出反应,以调节多种蛋白质的活性,包括SREBP和TFEB等转录因子。该网络是细胞活动的中央调节器,包括自噬、mRNA 和核糖体生物合成、蛋白质合成、葡萄糖、核苷酸和脂质代谢、线粒体生物合成和蛋白酶体活性。如果存在营养且压力较低,网络活动会通过激活合成代谢或通过诱导细胞防御途径来响应压力和营养短缺,从而对营养和压力状态做出反应。网络内及其与其他细胞内信号传导途径。,营养感应网络成分的基因活性降低可以延长寿命和健康寿命178、179(表1)。此外,遗传关联研究表明FOXO3 转录因子180和编码该网络组件的遗传变异178人类细胞的营养感应有关。181在青年时期,该信号网络的活动可促进有益的合成代谢过程,但在成年期间,它会获得促衰老特性(图 4)。

图4 .代谢改变
显示了一个简化版本的相互交织的营养途径,用于解除对营养感应的管制,并提供恢复营养感应的可能对策。值得注意的是,营养感应网络活性的降低会影响衰老过程中代谢调节以外的许多过程,包括对各种压力源的抵抗、修复机制的激活、自噬刺激或炎症控制。同样,对于线粒体功能障碍,列出了一系列与年龄相关的变化及其可能的解毒剂。与年龄相关的代谢改变的功能后果(其中一些与衰老的其他标志相关)在图的下半部分举例说明。
生长激素轴——历史上第一个与控制衰老有关的轴——是一种生长刺激级联反应,其顶点涉及垂体产生的生长激素(GH)。 GH 作用于肝细胞的GH 受体,刺激 IGF(特别是IGF1)的分泌, IGF1R 通过激活 PI3K-AKT 和MTORC1网络来刺激营养信号,从而促进生长和发育。 182在多种模式生物中,该途径的自发或工程突变可延长寿命并延缓与年龄相关的退化(表 1)。生长营养轴的先天缺陷会导致侏儒症,但从成年早期抑制该轴对机体健康具有有益影响(图 4)。
参与营养感应的另一个信号通路依赖于受体酪氨酸激酶 ALK(图 4),在小鼠中,该受体通过喂食183在下丘脑中被诱导,并对配体增强子 α 和 β(Augα 和 Augβ)做出反应。184在果蝇中,敲低 ALK 会降低甘油三酯水平和几种胰岛素样肽的表达,而遗传或药理学抑制 ALK 可延长健康寿命和寿命,尤其是在雌性中。183在小鼠中,全身或下丘脑特异性的 ALK 缺失以及 Augα 和 Augβ 的双敲除可促进对饮食引起的肥胖的抵抗力,而在人类中,ALK 的功能丧失突变与瘦身相关。183 , 184因此,该途径可能为干预代谢衰老提供额外的目标。
针对癌症和代谢疾病等疾病的药物通常会参与营养传感网络,因此此类药物是重新用作老年保护剂的候选药物。雷帕霉素和雷帕霉素类似物可破坏 MTORC1 复合物,已被证明可以延长模型生物体的寿命,即使在成年晚期开始治疗也是如此。185在小鼠中,雷帕霉素可以改善健康的各个方面,尽管它会加剧白内障等一些与年龄相关的特征,并且它在神经退行性疾病和其他与年龄相关的疾病模型中具有保护作用。
老年人容易受到病毒性呼吸道感染。用 MTORC1 抑制剂进行预处理可增强老年志愿者对流感免疫的免疫反应186并减少随后冬季的病毒性呼吸道感染187 ,从而指出了逆转与年龄相关的免疫衰老的潜在策略。
机制
在人类中,IGF1 在生命的第二个十年达到顶峰,但随着衰老而下降。在成年或晚年抑制 GH/IGF1 通路可延长模型生物体(包括小鼠)的寿命。48通过 PI3K 显性失活p110α 同工型的表达抑制心脏 IGF1R ,可延长雄性小鼠的最大寿命,并改善老年小鼠的心脏功能。188此外,用酪氨酸激酶抑制剂对 IGF1R 进行酶抑制可改善恶性细胞中需要诱导自噬的抗癌免疫监视。 189长期施用抗 IGF1R 抗体可延长雌性(而非雄性)小鼠的寿命,同时减少炎症和肿瘤的发展。这些发现表明 IGF1/IGF1R 信号轴可能构成抗衰老干预措施的目标。支持这一猜想的是,在老年女性(≥95 岁)以及老年人混合人群(平均年龄 76 岁)中,低 IGF1 水平与认知障碍和死亡的低概率相关。190此外,在英国生物银行的一个大型队列中,发现高 IGF-1 的风险与痴呆、糖尿病、血管疾病、骨质疏松症和总体死亡率的年龄之间存在显着的正相关性。191在百岁老人中,IGF1BP2 和 IGFBP6 的浓度升高。192未来将告诉我们,尚未开发的抗体或小分子是否可以选择性抑制 IGF1R 信号传导而不影响其他受体酪氨酸激酶(特别是胰岛素受体),可用于生长激素轴的脉动抑制以实现健康益处具有可接受的副作用。
营养的影响
饮食是干预人类衰老最实际的目标之一。从机制上讲,营养过剩:(1)触发细胞内营养传感器,如MTORC1(由亮氨酸和其他氨基酸激活)和乙酰转移酶EP300 (由乙酰辅酶A激活); (2) 抑制检测营养缺乏的传感器,例如 AMP 激活激酶 (AMPK) 和脱乙酰酶SIRT1和SIRT3(对 NAD +做出反应); (3) 消除分解代谢反应(糖原分解、糖异生的蛋白水解以及与生酮相关的脂肪分解),从而抑制适应性细胞应激反应,包括自噬、抗氧化防御和 DNA 修复。相反,禁食和饮食限制会抑制 MTORC1 和 EP300;激活 AMPK、SIRT1 和 SIRT3;并刺激适应性细胞应激反应,因为它们抑制生长激素轴并延长包括灵长类动物在内的多种模型生物的寿命。193
营养传感器构成了潜在的长寿药物的目标(图 4),但饮食限制也可能实现健康益处和延长寿命。从机制上讲,这可以通过减少总热量摄入、控制饮食成分194、195或限时喂养来实现。196如果雄性 C57BL/6J 小鼠在白天完全缺乏营养,饮食限制方案在延长雄性 C57BL/6J 小鼠的寿命方面特别成功。49然而,饮食限制方案并不能延长所有小鼠品系的寿命,这支持了饮食限制方案必须适应每个个体的基因组成的论点。197在人类中,基于饮食限制的临床检测因依从性差而变得复杂,但表明对免疫和炎症有积极作用。50
间歇性禁食(例如,1 天无营养,然后 1 天随意进食)可以避免热量限制引起的长期体重减轻,但可以延长小鼠的寿命195并改善临床试验中的健康生物标志物。198 , 199果蝇中类似的间歇性禁食方案的寿命延长归因于自噬刺激基因的夜间特异性上调,200但这尚未在哺乳动物中进行研究。雷帕霉素诱导的寿命延长(在果蝇中部分取决于自噬诱导)可以通过持续长期暴露以及间歇性治疗方案来获得,201表明该轴的脉冲抑制足以获得寿命延长的益处。尽管每 3-4 周进行 4-7 天的部分热量限制可能足以改善代谢综合征和抗癌免疫监视,但临床使用这种间歇性治疗的最佳间隔尚未确定。第202章
另一种潜在有益的疗法是生酮饮食,这是一种低碳水化合物、高脂肪和充足蛋白质的饮食。禁食和生酮饮食都会增加酮体(特别是 3-羟基丁酸)的产生,酮体是在肝脏中以自噬依赖性方式由乙酰辅酶 A合成的,可以在血浆中达到毫摩尔浓度并取代葡萄糖作为必需燃料例如,用于维持大脑功能。203在饮用水中永久但非循环施用 3-羟基丁酸酯可延长小鼠的寿命和健康寿命。 51这强烈表明这种酮体介导了生酮饮食的一些有益作用。从机制上讲,3-羟基丁酸诱导血管舒张并激活作用于 GTP 蛋白偶联受体 109A、203的免疫反应,而它直接抑制NLRP3 炎症小体204,表明潜在的多效性作用模式。
7、线粒体功能障碍
线粒体不仅是细胞的动力源,而且还构成炎症的潜在触发因素(当活性氧[ROS] 或 mtDNA 从细胞器中泄漏时,分别导致炎症小体或胞质 DNA 传感器的激活)和细胞死亡(当半胱天冬酶的激活剂、核酸酶或其他致命酶从膜间隙释放)。146随着衰老,线粒体功能因多种相互交织的机制而恶化,包括线粒体 DNA 突变的积累、蛋白质稳态缺陷导致呼吸链复合物不稳定、细胞器周转减少以及线粒体动力学变化。这种情况损害了线粒体对细胞生物能的贡献,增强了 ROS 的产生,并可能引发线粒体膜的意外透化,从而导致炎症和细胞死亡。 182从逻辑上讲,线粒体的功能对于维持健康至关重要,其逐渐退化会导致衰老表型(图 4)。
线粒体功能和寿命
延长健康寿命的干预措施可以刺激线粒体的功能。例如,安慰剂对照试验表明,补充左旋肉碱对体弱前受试者和老年男性都有积极作用57(表 1)。这种作用可能是通过抵消与年龄相关的左旋肉碱水平下降来介导的,这可能会限制线粒体对脂肪酸的氧化。 205矛盾的是,在模型生物体中,可以通过损害线粒体功能来延长寿命,线粒体功能会诱导毒物兴奋反应(“线粒体毒物兴奋作用”),前提是这种抑制是部分的并且发生在发育早期。在秀丽隐杆线虫中,部分抑制线粒体蛋白质合成或输入可通过涉及线粒体UPR (UPR mt )的机制延长寿命。 206在果蝇中,复合物 I 亚基 NDUFS1/ND75 的肌肉特异性敲除以UPR mt依赖性方式延长寿命。207用 TPP-噻唑 轻度抑制线粒体 ATP 合成可以改善衰老小鼠的代谢健康,减少内脏脂肪并改善葡萄糖耐量、线粒体质量和氧化代谢。52通过控释线粒体质子载体 (CRMP) 实现肝线粒体部分解偶联,还可逆转高脂饮食诱导肥胖小鼠的年龄相关代谢综合征。53在非人类灵长类动物模型中,包括自发肥胖的恒河猴和高脂肪、高果糖喂养的食蟹猴,CRMP 可逆转代谢综合征的迹象并改善脂肪酸氧化。 55这些效应与肝脏乙酰辅酶 A 水平的降低有关,这种现象已知会刺激自噬。208质子载体诱导线粒体自噬,209这也可以解释它们对新陈代谢的积极影响。二甲双胍是一种被认为是弱复合物 I 抑制剂的抗糖尿病药物,已被讨论为可能的抗衰老药物。210然而,到目前为止,没有证据表明挑战线粒体可以延长人类的健康寿命或寿命。
由于血清/糖皮质激素调节激酶 1 的缺乏而导致线粒体膜通透性 (MMP) 增加,从而缩短寿命,当自噬增强时,寿命会进一步受到损害,但当自噬被线虫中必需的自噬相关基因敲低所抑制时,寿命会恢复正常。211因此,MMP 可能构成一种危及生命的病症,并因自噬而加剧。一种修饰的四肽,elamipretide,已被开发用于靶向线粒体内膜(IMM)中的心磷脂,然后与IMM 蛋白腺嘌呤核苷酸转位子-1结合以抑制线粒体通透性转变,这是导致线粒体通透性转变的一种特殊机制。 MMMP。54 Elamipretide 对小鼠多种衰老相关表型具有积极作用,并在针对 Barth 综合征患者的临床试验中取得了积极结果56(表 1)。了解埃拉米普肽是否可以与其他延长寿命的药物(包括自噬增强剂)联合使用非常重要。除了这些工作之外,还有几项临床前和临床研究评估抗氧化剂亲脂性阳离子 MitoQ 和 SkQ1 的潜在有益作用。212进一步的研究将确定所有这些化合物在旨在改善与年龄相关的线粒体功能障碍的其他干预措施中的效用。
线粒体微生物蛋白与衰老
由线粒体DNA编码的微生物蛋白护脑素的血浆水平随着年龄的增长而下降。然而,百岁老人及其后代表现出高水平的护脑素。213值得注意的是,人类中的护脑素水平与IGF1呈负相关,使用GH或 IGF1 治疗 GH 不足的患者会减少循环护脑素。214 秀丽隐杆线虫中的护脑素转基因表达可通过自噬诱导延长寿命,并且用护脑素类似物 HNG 治疗中年小鼠可改善代谢健康寿命并减少全身炎症。213另一种线粒体 DNA 编码的微生物蛋白 MOTS-c 会随着年龄的增长而下降,但可以通过运动来诱导。 215 MOTS-c 有利于代谢物 5-氨基咪唑-4-甲酰胺-1-β-4-呋喃核苷 (AICAR) 的产生,它作为内源性AMPK 激动剂,从而预防年龄依赖性和高脂肪饮食引起的胰岛素抵抗以及饮食引起的肥胖。215因此,线粒体微生物蛋白作为潜在的抗衰老因子出现,将细胞器功能与机体稳态联系起来。
8、细胞衰老
细胞衰老是由急性或慢性损伤引起的反应。216在人类中,衰老细胞以不同的速度在多个组织中积累,与年轻(<35 岁)和老年(>65 岁)健康供体相比,衰老细胞的积累速度为 2 至 20 倍,217主要影响成纤维细胞、内皮细胞和免疫细胞尽管所有细胞类型都可能在衰老过程中经历衰老,218这一过程至少部分是由端粒随衰老缩短而触发的。109事实上,即使是有丝分裂后或缓慢增殖的组织,例如大脑或心脏,也可能含有衰老细胞。219此外,许多疾病中都会出现衰老细胞的局灶性或组织特异性积累。220关于细胞衰老在衰老中的因果作用最令人信服的证据是,持续通过基因或药物消除衰老细胞可以延长自然衰老小鼠的健康寿命和寿命。59此外,通过基因或药物消除衰老细胞可治疗许多小鼠模型疾病,221且至少有 3 项临床试验已完成,15 项临床试验正在进行或计划针对多种适应症的衰老。222
引发原发性衰老的损伤类型包括致癌信号、基因毒性损伤、端粒极短、线粒体损伤、病毒或细菌感染、氧化损伤、营养失衡和机械应激。216此外,继发性或旁分泌衰老可由细胞外炎症和纤维化介质(包括CCL2、IL-1β、IL-6、IL-8 和 TGF-β)触发。 223有证据表明,原发性衰老和继发性衰老在相关生物学方面存在差异,但这种区别的分子基础仍然难以捉摸。从历史上看,细胞衰老最显着的特征是由肿瘤抑制因子TP53和 CDKN2A/p16 及其下游效应子 CDKN1A/p21 和视网膜母细胞瘤-1 (RB1) 家族蛋白分别激活介导的稳定增殖停滞 。这些蛋白质共同抑制驱动细胞周期的细胞周期蛋白依赖性激酶 (CDK) 和转录激活剂(E2F 家族)。 216衰老过程中的另一个重要事件是核膜中核纤层蛋白B1的耗尽。这导致核纤层蛋白相关异染色质丢失,并从头形成富含 H3K9me3 的异染色质,这一过程可以可视化为 HP1α 灶或衰老相关异染色质灶 (SAHF)。224最终结果是长期且可行的增殖抑制,自发逃逸率较低。根据其分子组成,接受基因毒性治疗的癌细胞可能会经历典型的衰老反应,并具有高度稳定的细胞周期停滞,或者可能会经历类似衰老的反应,并具有高度可逆的停滞,甚至可以完全绕过衰老。225值得注意的是,衰老还在胚胎发生过程中特定细胞和结构的程序性消除中发挥着作用。 226
衰老与人类疾病
细胞衰老与多种非增殖性疾病有关,包括肺纤维化、肾脏疾病、肝脏脂肪变性、肥胖相关代谢综合征、I型和II型糖尿病、动脉粥样硬化以及阿尔茨海默病和帕金森病。220细胞衰老在这些疾病中的致病作用可以通过衰老相关分泌表型 (SASP) 来解释。 SASP源于衰老细胞的三个特征:(1)内源性逆转录病毒的转录去抑制,尤其是LINE-1,导致双链DNA胞质渗漏并激活cGAS/STING和TLR通路;136 (2) 线粒体ROS过量产生; (3) 自噬-溶酶体系统的扰动导致溶酶体含量增加,从而促进溶酶体衰老相关 β-半乳糖苷酶 (SABG) 的组织化学检测。227
SASP 具有高度异质性,具体取决于先天免疫信号通路(cGAS/STING、TLR 和 NLRP)、mTORC1 和转录因子(NF-κB、CBP、GATA4等)的细胞类型特异性激活。 SASP 通常对微环境产生同时且部分冲突的后果:(1)通过分泌趋化因子(CCL2、CXCL2和 CXCL3)和细胞因子(IL-1β、IL-2、IL-6 和IL-8); (2)通过分泌TGF-β来抑制免疫系统; (3)通过促纤维化因子(TGF-β、IL-11和PAI1)触发成纤维细胞活化和胶原沉积; (4)通过分泌基质金属蛋白酶来重塑ECM; (5)通过生长因子(EGF和PDGF)的分泌触发祖细胞的活化和增殖; (6) 触发邻近细胞的旁分泌衰老(TGF-β、TNF-α 和 IL-8)。在许多疾病中,SASP 的净效应是慢性炎症和进行性纤维化。228
尽管没有一个明确的细胞衰老标记,但可以通过多种特征组合的共存来识别这一过程,这些特征组合在一起是特定的,并为该现象提供分子定义:216 (1) 溶酶体扩张,可检测由 SABG 提供; (2)CDK抑制剂的上调,特别是p16和/或p21; (3) LMNB1从核膜中丢失; (4) 染色质成分HMGB1 从细胞核中丢失,并作为警报素释放到细胞外; (5) 异色病灶,可视化为 HP1γ 核病灶或 SAHF; (6) ROS水平高; (7) DNA 损伤加剧,表现为 γH2AX 核灶; (8) 高水平的 SASP 因子,特别是 IL-6、TGF-β、PAI1 等。
鉴于细胞衰老与多种病理学之间的关联,出现了关于这种细胞反应的生物学目的的问题。细胞衰老是一种有效的肿瘤抑制机制,但越来越多的证据已将细胞衰老与组织修复过程联系起来,在组织修复过程中,衰老细胞促进局部纤维化和免疫细胞的募集,然后清除受损和衰老的细胞。在这方面,组织修复可以被认为是一个两步过程:细胞衰老,随后是免疫招募和衰老的免疫清除(图5A)。在这种情况下,衰老是一种暂时受限的反应,可以通过自我消除来产生有益的结果。229只有当免疫清除的第二步没有实现时,衰老的病理后果才会变得可见,并且衰老细胞的积累和SASP对组织微环境的影响最终导致纤维化。

图5 .细胞衰老和干细胞耗竭
(A) 细胞衰老通常会促进损伤后的组织修复,并保护生物体免受致癌损伤。这是通过两个步骤实现的:(1) 衰老的建立;(2) 招募免疫细胞,消除衰老细胞,从而促进组织修复。如果这些步骤中的任何一个失败,生物体就很容易患上疾病。
(B) 干细胞衰竭是由于组织修复所需的细胞可塑性丧失所致。组织修复需要通过分泌细胞因子(部分由于衰老相关的分泌反应)、生长因子和细胞外基质(ECM)调节剂来改变微环境,有利于来自不同组织区室的细胞的去分化和可塑性。这些损伤诱导的塑性细胞可能获得多能祖细胞功能。 OSKM因子的瞬时表达会抑制细胞身份程序的转录,从而导致整体去分化(OSKM on)和可塑性的获得。为了恢复活力,必须在此时中断该过程(OSKM关闭),以允许细胞重新分化并恢复其原始细胞身份。
延年益寿药
细胞衰老与多种病理学之间的密切联系促使人们寻找能够选择性杀死衰老细胞的小化合物,这些化合物被称为“senolytics”。230 ”值得注意的是,衰老作用(消除衰老细胞)与衰老反应的取消有很大不同,后者可能是由 p16 或 p21 突变等引起的。衰老作用不会阻止衰老的执行,而是概括衰老细胞的自然免疫清除(图 5 A)。为了支持这一点,长期接受遗传诱导或药物诱导的衰老作用的小鼠表现出更长的寿命,而没有增加癌症发病率或有缺陷的组织修复迹象。59 , 58
senolytic 疗法的数量仍然有限,但有些已广泛用于疾病的临床前模型,例如 navitoclax、达沙替尼和槲皮素(D/Q) 双重治疗、非瑟汀、强心苷等。221衰老细胞的存活和抗凋亡能力在很大程度上取决于 BCL2 蛋白家族,特别是 BCLXL,但也依赖于 BCL2 和 BCLW。这使得衰老细胞极易受到针对这三种蛋白质的 navitoclax 的影响。 231 Navitoclax 已在临床试验中进行了抗肿瘤活性评估,预计该药物(或对血小板无毒性的衍生物)将进入衰老相关疾病的临床试验。232其他潜在的 senolytic 治疗方法,如 D/Q 230和 fisetin 60已被批准用于人类,并且正在针对多种适应症进行各种临床试验中进行测试。它们作用的机制基础仍不清楚。达沙替尼是一种混杂激酶抑制剂,槲皮素和非瑟酮是具有多个靶点的天然黄酮类化合物。 D/Q 已在临床试验中进行了测试,在肺和肾纤维化的情况下取得了有希望的结果。62 , 61强心苷抑制所有细胞中存在的质膜 Na + /K + -ATP 酶,导致阳离子失衡并降低细胞内 pH 值。233强心苷的衰老作用机制可能与衰老细胞对低细胞内 pH 值的脆弱性有关。因此,谷氨酰胺酶的化学抑制剥夺了细胞抵抗低pH值的机制并导致衰老作用。234所有上述讨论的衰老化合物在多种与衰老相关的小鼠疾病模型中发挥治疗活性。衰老作用也可以通过针对衰老细胞表面出现的蛋白质的免疫学方法来实现。特别是,针对糖蛋白NMB (GPNMB) 235的抗体和针对受体 uPAR 236 的CAR T 细胞可减轻小鼠衰老相关疾病模型。
总之,细胞衰老是对应激和损伤的重要反应,在正常生理学中,随后是免疫清除,但在衰老或慢性损伤时,免疫机制无法消除细胞衰老,因此由于前体细胞的大量分泌而具有致病性。 -炎症和促纤维化因子。旨在杀死衰老细胞的治疗策略已在动物模型中进行了广泛探索,目前正在进行临床试验(表1)。
9、干细胞衰竭
衰老与稳定状态下的组织更新减少以及受伤时组织修复受损有关,每个器官都有自己的更新和修复策略。237例如,在骨骼肌中,一种单细胞类型(卫星细胞)被放置在单能和单向层次结构的顶端,用于更新和修复。皮肤表皮的特点是高度更新和容易受伤,有多个干细胞生态位,特别是与毛囊相关的干细胞生态位,每个干细胞生态位都会产生其后代和区域。然而,在受伤后,多个细胞可以获得干细胞特性并颠覆领域边界。其他器官,如肝脏、肺或胰腺,在正常条件下表现出相当低的更新率,这与不同细胞类型获得的干细胞特性(包括增殖和多能性)形成鲜明对比(图 5 B)。事实上,组织修复被认为在很大程度上依赖于损伤诱导的细胞去分化和可塑性。例如,在肠、脑和肺中,损伤会诱导非干细胞去分化,从而重新激活通常沉默的胚胎和干细胞转录程序,从而获得组织修复所需的可塑性。238 , 239 , 240损伤引起的可塑性(及其随着衰老而逐渐丧失)可能比正常稳态条件下驻留干细胞的可塑性与衰老更相关。干细胞和祖细胞与没有干细胞潜力的细胞一样,都具有相同的衰老特征,因此,我们在此不讨论有关每种衰老特征对干细胞功能影响的大量文献。相反,我们将基于“细胞重编程”的概念,重点关注对抗干细胞功能随衰老而下降的总体策略。该过程被认为以细胞自主的方式作用于多种细胞类型。然而,由于其长期影响,它对干细胞和祖细胞的影响被认为具有更高的相关性。
通过重编程使组织修复恢复活力
细胞向多能性重编程包括通过四种外部转导转录因子(即 OCT4、 SOX2、KLF4和 MYC (OSKM))的同时作用,将成体体细胞转化为胚胎多能细胞(称为诱导多能干细胞或 iPSC)。 。 241重编程过程通常需要几周的时间,在此期间,细胞首先通过细胞识别基因的转录抑制而失去其分化表型,随后反式激活多能性基因。 242完全重编程不仅意味着细胞身份的改变,还意味着细胞的复兴,其特征是许多衰老特征被重置为胚胎状态,如p16减少、端粒延长、 244和 DNA甲基化时钟重置所示。。 245有趣的是,在去分化开始后不久,复兴就以渐进的方式发生。 246事实上,可以使用 OSKM 启动重编程,在中间状态中断该过程,并允许细胞返回其原始身份。这种短暂的细胞扰动,被称为“部分”、“短暂”或“中间”重编程,能够使衰老的细胞标志物恢复活力,例如DNA 甲基化时钟、DNA 损伤、表观遗传模式以及与衰老相关的细胞因子变化。转录组,体外和体内。 63,64,70,246,247因此,可以提出去分化和再生过程是耦合的。具体来说,去分化意味着表观遗传和转录程序的消除,这也可能消除与衰老相关的改变。部分重编程中断后,细胞在再分化过程中重新建立其原始的表观遗传和转录状态,有趣的是,它不会重新建立被消除的与衰老相关的变化,因此将表观基因组和转录组重置为更年轻的状态。
小鼠的瞬时重编程赋予了衰老组织的修复能力,因此随后的损伤可以像年轻个体一样有效地修复。这种增强的修复能力已在内分泌胰腺、63骨骼肌、63神经纤维、70眼睛、70皮肤、64心脏、65和肝脏等组织损伤模型中得到证实。 66此外,自然衰老所特有的组织功能障碍,例如视力下降70以及海马体中成人神经发生和长期记忆的丧失67可以通过瞬时重编程部分逆转。在某些情况下,短暂重编程在组织修复过程中也是有益的(而不仅仅是在受伤之前)。这是脑外伤68和皮肤伤口愈合的情况。69最后,应该提到的是,早衰小鼠的寿命可以通过瞬时重编程来延长,63尽管尚未有 OSKM 延长野生型小鼠寿命的报道。
部分重编程概括了自然组织修复的特征(图 5 B)。在这两种情况下,细胞都会经历短暂的去分化过程、获得胚胎和祖细胞特征以及随后的再分化。因此,去分化和再分化可以解释组织再生,这与肌细胞短暂去分化随后再分化诱导 转录组再生的观察结果一致。248组织修复的自然过程可能意味着某种程度的细胞再生,这与表观遗传甲基化时钟在组织损伤后很快加速并在组织修复过程中部分逆转的发现一致。249此外,据报道,组织损伤创造了一个高度允许 IL-6 驱动的重编程的组织微环境。250最后,转录因子 FOXM1的循环表达延长了早衰小鼠和野生型小鼠的寿命。251虽然详细机制尚未探索,但FOXM1在损伤后在肾脏中被诱导,并在修复过程中参与触发肾小管上皮细胞的去分化和增殖。252因此,自然组织修复和人工重编程的几个特征可能会融合,也许可以改进恢复老化组织修复能力的策略。
10、改变细胞间通讯
衰老与细胞间通讯的逐渐改变有关,从而增加系统中的噪音并损害体内平衡和毒物兴奋调节。因此,衰老涉及神经、神经内分泌和激素信号传导途径的缺陷,包括肾上腺素能、多巴胺能、胰岛素/IGF1系统和肾素-血管紧张素系统,以及与生殖功能丧失相称的性激素。182 , 253虽然这种改变的主要原因是细胞内在的,因为这在 SASP 中得到了特别详细的记录,但细胞间通讯的这些紊乱最终总结为其自身的标志,将细胞内在标志与元细胞标志联系起来包括炎症反应的慢性化以及针对病原体和癌前细胞的免疫监视能力的下降,以及人类基因组和微生物组之间双向通讯的改变,最终导致生态失调。这方面的许多研究都集中在寻找具有促衰老或延长寿命特性的血源性全身因子、细胞间不同通讯系统的作用以及评估衰老过程中 ECM 破坏的功能相关性。
促衰老血源性因素
单次输注老年血液会在几天内引起年轻小鼠的衰老特征,72,并且用含有 5% 白蛋白的生理盐水缓冲液简单稀释老年小鼠的血液会诱导多个组织的再生,71表明循环因子的存在有利于衰老过程。在促衰老血源性因子中, 趋化因子CCL11/eotaxin 和炎症相关蛋白 β2-微球蛋白会减少神经发生,254、255 IL -6 和 TGF-β 会损害造血功能,256补体因子C1q会损害肌肉修复。257理论上,这些因素的中和可能具有有效的抗衰老作用。事实上,上述几个因素是在 SASP 的背景下分泌的,可能是“传染性”衰老现象的共同原因,这种现象也涉及细胞外囊泡。258因此,所谓的“senomorphics”可能用于抑制 SASP 并延缓衰老。
抗衰老血源性因子
年轻小鼠血液中存在的可溶性因子可有效恢复年老小鼠的更新和修复能力259(表1)。异时性联体共生实验以及广泛的单细胞转录组学证实了年轻血液具有使多个组织恢复活力的能力74并恢复与年龄相关的一般基因表达的减少,特别是参与电子传递链的线粒体基因的减少。75趋化因子 CCL3/MIP-1α 作为造血干细胞和祖细胞的再生因子;74金属蛋白酶抑制剂TIMP2与海马体的恢复活力有关;73抗炎白细胞介素IL-37(老年人的单核细胞数量减少)可提高老年小鼠的耐力运动并改善全身代谢;76细胞因子GDF11可以使一些组织恢复活力,例如肌肉、大脑和内分泌胰腺,但由于其促纤维化副作用,会损害其他组织的功能和修复;77最后,转基因强制 VEGF 过度表达的小鼠表现出肝脏和肌肉修复增强、总体健康状况改善以及平均寿命延长约 40%。78
远程和短程通信系统
中枢神经系统控制影响外周器官的衰老的多个方面,解释了大脑特异性基因操作(例如 SIRT1、UCP1 的过度表达或 IKBKB 和 TRPV1 的敲除)如何能够延长小鼠的寿命(表 1)。这些长期活动的确切机制尚未确定。260值得注意的是,细胞间通讯还涉及短寿命细胞外分子(例如 ROS、一氧化氮、核酸、前列腺素和其他亲脂性分子)、从各种组织(包括白色脂肪组织)释放的可溶性因子(脂肪因子)之间的相互作用。 、棕色脂肪组织(baptokines)、心脏(cardiokines)、肝脏(hepatokines)和骨骼肌(肌因子,包括运动产生的运动因子)、细胞结合配体和其他细胞上的受体(例如IL-1α,可以保持细胞结合),以及由紧密连接或间隙连接介导的直接细胞间相互作用。所有这些通信系统都可能在衰老过程中发生改变,因此正在仔细检查它们潜在的促衰老和抗衰老特性。258
细胞外基质
衰老会对 ECM 的长寿命蛋白质成分造成多种损害,包括AGE、羰基化和氨甲酰化、弹性蛋白断裂和胶原交联,261从而导致组织纤维化(纤维化)。 262这种有害过程的部分原因是 TGF-β 和其他生长因子的过度释放,以及 TAZ 和 YAP 转录因子的核转位,这些转录因子充当机械转导器并触发促纤维化基因(例如转谷氨酰胺酶 2)的表达、赖氨酰氧化酶(LOX) 和 LOX 样酶。262 ECM 硬度还会影响衰老细胞的功能,衰老细胞反过来会分泌基质金属蛋白酶,放大 ECM 的损伤, 263并通过蛋白水解产生损伤相关分子模式,以激活促衰老、促纤维化和促炎症途径。 262老化基质的硬度增加也可能有利于 WNT 信号传导,诱导成纤维细胞活化和促纤维化基因表达。 264该通路与其他促纤维化通路(例如 NOTCH、RAS、TGF-β/SMAD 和 Hedgehog/GLI)表现出广泛的串扰,从而证明了年龄相关纤维化发展机制的复杂性和相互关联性。 262值得注意的是,在机械响应离子通道 PIEZO1 介导的过程中,基质刚度引起的机械变化足以导致少突胶质细胞祖细胞与年龄相关的功能丧失。 265
多项研究为 ECM 硬度对衰老的影响提供了因果证据,并提出了改善健康衰老的方法(表 1)。使用AAV 载体体内抑制 Piezo1可使年老小鼠大脑中的少突胶质细胞祖细胞恢复活力。265基质细胞中 YAP/TAZ 的基因失活会导致加速衰老,尽管维持 YAP 功能可以使衰老细胞恢复活力,并通过控制 cGAS-STING 信号传导来防止衰老特征的出现。79此外,经过基因改造以产生抗胶原酶I 型胶原(Col1a1r/r) 的小鼠表现出血管细胞衰老、加速衰老和寿命缩短。266 COL25A1(编码大脑特异性胶原蛋白)的罕见变体的发现进一步强化了胶原蛋白对人类长寿的重要性,这种变体可能对阿尔茨海默氏病具有保护作用。 267此外,由年轻的人类成纤维细胞制备的 ECM 可诱导衰老的衰老细胞进入年轻状态。硫酸软骨素和透明质酸等80 种ECM 化合物可恢复与年龄相关的胶原蛋白下降,并延长线虫的寿命。268相反,人透明质酸酶TMEM2的异位表达可通过 p38/ERK MAPK 信号传导的变化促进线虫对ER 应激的抵抗并延长寿命。269在小鼠中,软骨素6-磺基转移酶的缺失会导致大脑中的 ECM 异常、早期记忆丧失并加速大脑衰老,而这种酶的过度表达可改善老年小鼠的记忆力。81回顾性分析表明,口服葡萄糖胺/硫酸软骨素可降低人类全因死亡率。270然而,迄今为止还没有前瞻性证据表明这种延长效应可以通过改善 ECM 来调节。
11、慢性炎症
衰老过程中炎症会增加(“炎症”),并伴有全身表现以及局部病理表型,包括动脉硬化、神经炎症、骨关节炎和椎间盘退变。因此,炎症细胞因子和生物标志物(例如 CRP)的循环浓度随着衰老而增加。血浆中升高的 IL-6 水平构成老龄化人群全因死亡率的预测生物标志物。271与炎症加剧相关的是,免疫功能下降,这种现象可以通过对患者和小鼠组织血液中的骨髓细胞和淋巴细胞进行高维监测来捕获。 272例如,一群与年龄相关的 T 细胞(称为 Taa 细胞)由耗尽的记忆细胞组成,通过颗粒酶K 介导促炎作用。T细胞群的转变导致促炎 TH1 和TH17 细胞功能亢进,免疫监视缺陷(对消除病毒感染的恶性或衰老细胞产生负面影响),自我耐受性丧失(随之而来的是与年龄相关的自身免疫性疾病的增加),以及生物屏障的维护和修复减少,总体上有利于全身炎症273(图6A)。

图6 .超细胞功能紊乱
细胞间通讯的改变将细胞内在特征与元细胞特征联系起来,包括慢性炎症,以及人类基因组和微生物组之间串扰的改变,最终导致生态失调。
(A) 衰老过程中的慢性炎症是由所有其他特征引起的多种紊乱的结果。图中右侧部分显示了抗炎干预措施对健康和寿命产生积极影响的几个代表性例子。
(B) 生态失调会导致多种与衰老相关的病理状况。人类肠道微生物群在衰老过程中发生显着变化,最终导致生态多样性普遍下降。右图的下半部分显示了这些微生物群变化背后机制的主要特征,以及对肠道微生物群组成进行干预以促进健康衰老的一些例子。 CVD、心血管疾病;SCFA,短链脂肪酸。
炎症与其他衰老特征之间的联系
炎症的发生是由所有其他特征引起的多种紊乱的结果。例如,炎症是由细胞核和线粒体 DNA 易位进入细胞质而引发的,在细胞质中刺激促炎 DNA 传感器,特别是当自噬无效并因此无法拦截异位 DNA 时。4基因组不稳定性有利于不确定潜能克隆造血(CHIP),伴随着通常具有促炎表型的骨髓细胞的扩张,导致心血管衰老等。274有趣的是,最常见的 CHIP 相关突变影响表观遗传修饰剂 DNMT3(甲基化DNA 中的胞嘧啶残基)和 TET2(催化甲基胞嘧啶氧化为 5-羟甲基胞嘧啶)。从机制上讲,影响 TET2 的 CHIP 会增强骨髓细胞产生 IL-1β 和 IL-6,并刺激心血管疾病 (CVD),而这种疾病在 IL-6 受体功能丧失突变或接受 IL-6 受体治疗的个体中会减弱。 1β中和抗体。275
促炎蛋白的过度表达可能继发于表观遗传失调、蛋白质稳态缺陷或自噬功能障碍。过多的营养信号导致 GH/IGF1/PI3K/AKT/mTORC1 轴激活,从而引发炎症。此外,继发于衰老细胞累积的SASP、细胞外碎片和感染性病原体的积累以及由于衰老而未被清除的细胞外碎片和感染性病原体的积累,以及骨髓和淋巴样细胞的耗竭,都促进了炎症。后一种现象涉及与年龄相关的胸腺退化、胸腺生成功能的丧失以及随之而来的 T 细胞库的稀疏以及无法对新抗原产生有效的免疫反应。276值得注意的是,CR 可以改善人类的胸腺生成,并且可以在小鼠体内敲除编码血小板活化因子乙酰水解酶A2 VII 组 (PLA2G7)的 CR 下调基因,以对抗胸腺萎缩。50最后,昼夜节律紊乱和肠道屏障功能障碍也会加剧炎症。第277章
抗炎、抗衰老干预措施
尽管全身炎症在机制上与所有上述与年龄相关的改变有关,但炎症本身就构成了一个标志。事实上,对炎症和免疫系统的特定控制可以加速或减缓不同器官系统的衰老过程。例如,线粒体转录因子 A (TFAM) 中的 T 细胞特异性缺陷足以驱动心血管、认知、代谢和身体衰老,并伴有循环细胞因子的增加。 TNF-α 抑制剂依那西普部分逆转了这种表型。82小鼠造血细胞中DNA 修复蛋白ERCC1的杂合缺失 足以诱导非淋巴器官的免疫衰老和衰老,以及导致寿命缩短的众多器官损伤迹象。这种表型可以通过抗衰老非瑟酮缓解。278这些结果支持免疫系统衰老可能导致机体衰老的观点。值得注意的是,将TFAM缺失的 T 细胞、年轻的 ERCC 缺陷型脾细胞或衰老的野生型脾细胞过继转移到年轻小鼠体内会诱导衰老,而将年轻的免疫细胞转移到 ERCC 缺陷型小鼠体内会减弱衰老,这表明免疫细胞以积极和消极的方式调节机体衰老。82 , 278
有多个例子表明抗炎治疗可延长健康寿命并延长寿命(图 6A;表 1)。因此,阻断 TNF-α 可预防小鼠肌肉减少症并改善衰老大鼠的认知能力。83 , 84阻断常见1 型干扰素受体 (IFNAR1) 可逆转衰老小鼠肺部单核细胞的积累。 279敲除骨髓细胞中的前列腺素 E 2受体 EP2 或用药物 EP2 抑制剂治疗老年小鼠可改善认知能力。85敲除炎性体蛋白 NLRP3可改善代谢生物标志物、葡萄糖耐量、认知和运动性能,并延长小鼠寿命。86 NLRP3或其下游酶 caspase-1 的药理学抑制剂对正常和加速衰老模型具有令人鼓舞的临床前作用。 280最重要的是,用卡那单抗抑制 caspase-1 产物 IL-1β是适用于患者的抗衰老治疗的例证。 3 期临床试验 CANTOS 评估了canakinumab预防有心肌梗塞病史和明显炎症迹象的患者复发 CVD的能力。除了达到试验的主要终点外,卡那单抗还降低了糖尿病和高血压的发病率,以及肺癌的发病率和死亡率。87最后,尽管长期使用阿司匹林等非甾体类抗炎药可能对人类健康产生积极影响,特别是在预防心血管疾病和胃肠道癌症方面,但一项大型 3 期临床试验表明,阿司匹林在对 70 岁以上的受试者进行治疗,结果呈阴性。281因此,有必要进一步研究探索年轻时使用阿司匹林预防性治疗的价值,将阿司匹林与其他药物联合使用或用毒性较小的抗炎药替代阿司匹林。
12、生态失调
近年来,肠道微生物组已成为多种生理过程中的关键因素,例如营养消化和吸收、对病原体的保护以及必需代谢物(包括维生素、氨基酸衍生物、次生胆汁酸和短链脂肪酸 (SCFA))的产生。肠道微生物群还向外周和中枢神经系统以及其他远处器官发出信号,并强烈影响宿主健康的整体维持。146这种细菌-宿主双向通讯的破坏会导致菌群失调,并导致各种病理状况,如肥胖、2型糖尿病、溃疡性结肠炎、神经系统疾病、心血管疾病和癌症。282该领域的进展引起了人们对探索衰老过程中肠道微生物群改变的极大兴趣(图6B)。
衰老过程中的微生物群改变
由于宿主遗传变异(种族)、饮食因素和生活习惯(文化)以及环境条件(地理),肠道内的微生物群落在个体之间变化很大,这使得难以揭示微生物群与多效性年龄相关疾病表现之间的关系。尽管如此,一些荟萃分析揭示了微生物群与疾病的关联,这些关联已在不同的病理学中得到验证283和国家。284,285对人类和动物模型的研究提供了有关临床、流行病学、社会学和分子方面的宝贵信息,这些方面是老年微生物组对人类健康和疾病的复杂影响的基础。286一旦细菌多样性在儿童时期建立,在成年期保持相对稳定。然而,这种细菌群落的结构和活动在衰老过程中逐渐发生变化,最终导致生态多样性普遍下降。因此,对百岁老人种群进行的几项研究表明,核心丰富的分类群(如拟杆菌门和罗斯布里亚)有所减少,但几个属(如双歧杆菌属和阿克曼西亚属)也有所增加,这些属似乎具有延长寿命的作用。287
最近对来自三个独立队列的 9,000 多名 18-101 岁的个体的肠道微生物组和表型数据进行了分析,从而扩展了这些研究。288 值得注意的是,随着年龄的增长,个体肠道微生物组对每个人来说变得越来越独特,这种独特性与参与免疫调节、炎症和衰老的众所周知的微生物代谢物有关。在年龄较大时,健康的参与者表现出向独特微生物组成的持续漂移,而这种漂移在健康状况较差的个体中减少或不存在。已确定的健康老龄化微生物组模式的特征是核心分类群(如拟杆菌)的枯竭,这些类群存在于大多数人类中。此外,在接近极端年龄的个体中,高拟杆菌水平的保留和低肠道微生物组独特性测量与生存率降低显着相关。然而,来自百岁老人和超级百岁老人的微生物群的发现并不总是与来自老年人群的发现一致。ELDERMET研究报告称,与年轻对照组相比,核心属拟杆菌属、拟杆菌属和副拟杆菌属在老年个体中的优势性增加。这项研究还确定了与虚弱、认知、抑郁和炎症相关的肠道微生物群组成的年龄相关变化。289另一项研究揭示了不同种族人群中共享的微生物群的年龄相关轨迹,以及肠道微生物群中与性别依赖性差异的共同年龄相关减少。值得注意的是,老年人表现出更丰富的几种促进健康的细菌物种,包括阿克曼西亚。290这些结果表明,除了老年人的饮食变化和生活方式之外,与年龄相关的生理变化可能对人类肠道微生物群产生深远的影响。
所有这些研究结果的异质性表明,衰老可能存在多种肠道微生物组轨迹。然而,微生物群产生的氨基酸衍生物的血浆浓度存在有趣的趋同。这些代谢物包括源自色氨酸肠道细菌降解的吲哚,以及苯丙氨酸/酪氨酸的几种发酵产物,如对甲酚硫酸盐、苯乙酰谷氨酰胺和对甲酚葡糖苷酸。这一发现与ELDERMET队列的数据一致,该队列显示粪便中对甲酚的浓度与虚弱增加有关,并可能导致该人群与年龄相关的下降。相反,某些吲哚代谢物的血浆浓度与老年人健康状况的改善相关。吲哚代谢物至少部分地通过结合芳基烃受体减弱炎症反应来增加小鼠的健康寿命和寿命。93
对百岁老人肠道微生物组的进一步代谢组学和功能分析表明,它在某些特定细菌中富集,例如 Alistipes putredinis 和 Odoribacter splanchnicus。其中一些细菌物种能够产生独特的次生胆汁酸,包括异体胆汁酸,它对革兰氏阳性多重耐药病原体(如艰难梭菌和屎肠球菌)具有强大的抗菌作用。291因此,特异性胆汁酸代谢可能参与降低病原体感染的风险并有助于肠道稳态,从而降低对与年龄相关的慢性疾病的易感性。
粪便微生物群移植与衰老
病理衰老的多组学研究表明,两种不同的早衰小鼠模型表现出肠道菌群失调,主要特征是变形菌和蓝细菌丰度增加以及疣微生物水平降低。与这些发现一致,患有HGPS或NGPS的人类早衰症患者也表现出肠道生态失调,而长寿的人类表现出变形菌的显着减少和疣状微生物的显着增加。88这些变化的因果关系已通过粪便微生物群移植(FMT)在体内得到证实。在两种加速衰老模型中,从野生型到早衰小鼠接受的FMT都增强了健康寿命和寿命,而疣状微生物Akkermansia muciniphila的施用也足以获得这种效果。相反,从早衰供体到野生型受体的 FMT 诱导有害的代谢改变。恢复早衰小鼠中耗尽的次级胆汁酸和其他代谢物表型复制了重建健康微生物组的有益效果88 (表1)。
FMT还揭示了肠道菌群失调在慢性全身炎症以及与衰老和年龄相关疾病相关的适应性免疫下降中的致病作用。肠道微生物群从老年小鼠转移到年轻无菌小鼠引发了炎症反应,其特征是脾脏中CD4 T细胞分化增强,炎症细胞因子上调,细菌来源的炎症因子循环增加。+292FMT还为肠道微生物群在衰老过程中维持大脑健康和免疫力的影响提供了证据。90 来自年轻小鼠供体的微生物群逆转了海马代谢物和脑免疫的衰老相关差异,并改善了移植到老年宿主时认知行为的年龄相关障碍。这些工作开启了用益生元、益生元和后生元操纵肠道微生物群的可能性,以恢复免疫系统和衰老的大脑。异时粪便转移证实了微生物组成的年龄依赖性变化与宿主免疫系统功能下降之间的因果关系。92 事实上,老年小鼠Peyer斑块中的生发中心反应缺陷可以通过FMT从年轻动物身上拯救出来,而不会影响外周淋巴结中的生发中心反应。最后,来自年轻供体小鼠的FMT改善了老年小鼠的卵巢功能和生育能力。这些有益作用与老年卵巢免疫微环境的改善有关,巨噬细胞和巨噬细胞衍生的多核巨细胞减少,促炎IFNγ水平降低,抗炎细胞因子IL-4丰度增加。91
其他对肠道微生物群的延长寿命干预措施
益生菌植物乳杆菌 GKM3 在加速衰老的 SAMP8 小鼠模型中促进长寿并缓解与年龄相关的认知障碍。89 对肠道微生物群组成的干预也恢复了与年龄相关的小胶质细胞成熟和功能下降,这会导致大脑可塑性改变并促进神经退行性变。再定植实验或肠道微生物群代谢物(如短链脂肪酸)的施用可防止有益双歧杆菌的年龄相关下降,增加Akkermansia丰度,并恢复中年小鼠的小胶质细胞功能。94此外,热量限制饮食会引起肠道微生物组的结构变化,增加乳酸菌和其他影响健康衰老的物种的丰度。肠道微生物群诱导的炎症和随之而来的胰岛素抵抗增加也可以通过恢复老年小鼠和猕猴中有益的产生短链脂肪酸的细菌(如嗜粘液杆菌)的丰度来逆转。293同样,一项针对超重/肥胖胰岛素抵抗志愿者的随机、双盲、安慰剂对照试验研究表明,口服巴氏杀菌的嗜粘性曲霉可改善胰岛素敏感性,降低胰岛素血症和血浆总胆固醇水平。95总的来说,这些结果强调了衰老与生态失调之间的因果关系,并表明旨在恢复年轻微生物组的干预措施可能会延长健康寿命和寿命。
找助理微信号: erquzhuli 免费获取136页2024年最新版《蓝图计划》吧!
后记:深圳二区健康生命科技有限公司是一家以人工智能驱动,遵从循证医学、精准医学、再生医学理念的抗衰老长寿科技公司。公司将人工智能创新技术与生物医学相结合,致力于为生命健康领域的探索和进步做出贡献。
目前二区健康已有20+精准的健康长寿社群,欢迎添加助理微信号: erquzhuli,和我们一起来探索抗衰老长寿健康人生吧。